Fig. 12.1
Representative CT image demonstrating marked diffuse dilation of the small bowel without transition point
Introduction
Intestinal pseudo-obstruction can be categorized as either acute or chronic. Chronic intestinal pseudo-obstruction (CIP) differs clinically from acute intestinal pseudo-obstruction by the presence of obstructive symptoms for at least 6 months (Table 12.1). CIP is a rare yet debilitating neuromuscular disorder of the gastrointestinal tract characterized by impaired peristalsis with symptoms and imaging that may mimic mechanical bowel obstruction. Because the symptoms of CIP including abdominal pain, nausea, vomiting, bloating, and abdominal distension are nonspecific, CIP often goes undiagnosed for many years despite multiple potentially dangerous diagnostic tests and treatments. Thus, a high degree of clinical suspicion in conjunction with a careful history and physical examination remains paramount to establishing the diagnosis. Importantly, CIP is not a single disorder; rather, it refers to a heterogeneous group of disorders characterized by disordered intestinal peristalsis. Once mechanical obstruction is ruled out, dedicated imaging and motility testing can be utilized to help confirm the diagnosis. Unfortunately, a cure does not exist for CIP, and supportive care remains the cornerstone of disease management. Many patients with CIP have difficulty maintaining their normal weight and achieving adequate nutrition; a large percentage of CIP patients will eventually require nutritional support. For patients who fail symptomatic treatment or develop severe side effects from parenteral nutrition, small intestinal transplant has become a realistic treatment option.
Table 12.1
Definition of chronic intestinal pseudo-obstruction
The following must be present | Dilation of small intestine on radiography |
Symptoms of obstruction for longer than 6 months | |
The following must be absent | Mechanical obstruction |
Supporting studies | Delayed scintigraphy (i.e., gastric emptying scan) |
Abnormal esophageal or antroduodenal manometry | |
Connective tissue disease serologies (i.e., anti-scl-70) | |
Abnormal full-thickness biopsy of the small bowel |
Epidemiology and Natural History
CIP is a rare disorder. One estimate from a pediatric tertiary care center suggests that approximately 100 infants are born with CIP each year in the United States. The natural history for most CIP patients is that of a progressive worsening of their condition. In a recent report, the diagnosis of CIP was made a median of 8 years after symptoms first developed, and, during this time, each patient underwent an average of three surgeries related to their yet to be diagnosed CIP symptoms.
The long-term prognosis of CIP patients is poor. It is estimated that up to two-thirds of CIP patients develop a nutritional deficiency and that 30–50 % of adult CIP patients will require parenteral nutrition or small bowel transplantation. Many patients become opioid dependent due to chronic abdominal pain. CIP in the pediatric population has a similarly poor prognosis with a 10–25 % mortality rate before reaching adulthood.
Pathophysiology
The etiology of primary CIP is varied but may be characterized as a neuropathy, myopathy, or mesenchymopathy (i.e., affecting the interstitial cells of Cajal) depending upon the gut wall structure most affected. On the basis of these pathogenic abnormalities, a variety of both primary and secondary causes of CIP have been described. Secondary causes of CIP include but are not limited to collagen vascular disorders, endocrine disorders, neurologic disorders, and medications (Table 12.2). One of the more common secondary causes of CIP is primary systemic sclerosis. Importantly, certain malignancies (e.g., small cell carcinoma of the lung) may cause a paraneoplastic form of CIP. Irrespective of the etiology, the end result is that of impaired peristalsis and a poorly or nonfunctioning GI tract.
Table 12.2
Secondary causes of chronic intestinal pseudo-obstruction
Collagen vascular diseases |
|---|
Primary systemic sclerosis |
Systemic lupus erythematosus |
Dermatomyositis/polymyositis |
Periarteritis nodosa |
Rheumatoid arthritis |
Mixed connective tissue disease |
Endocrine disorders |
Diabetes |
Hypothyroidism |
Parathyroidism |
Neurologic disorders |
Parkinson’s disease |
Alzheimer’s disease |
Shy-Drager |
Chagas’ disease |
Intestinal hypoganglionosis |
Dysautonomia (familial or sporadic) |
Medication associated |
Tricyclic antidepressants |
Anticholinergic agents |
Ganglionic blockers |
Antiparkinsonian agents |
Clonidine |
Phenothiazines |
Miscellaneous |
Celiac disease |
Paraneoplastic syndromes (small cell lung carcinoma, carcinoid, thymoma) |
Infiltrative disorders (amyloidosis, lymphoma) |
Alcohol abuse |
Post-infectious processes (viral, bacterial, parasitic) |
Radiation |
Vascular insufficiency |
Metabolic (hypokalemia, hypomagnesemia) |
Postsurgical |
Post-organ transplant |
Mitochondrial disorders |
Diagnosis
The diagnosis of CIP may be elusive for a number of reasons. First, the symptoms do not typically develop at once but rather slowly evolve over a number of years. Second, CIP may affect all segments of the GI tract resulting in a variety of symptoms. Third, these symptoms which include abdominal pain, bloating, distension, nausea, vomiting, constipation, and weight loss are nonspecific. Fourth, initial diagnostic tests (see below) are usually normal. Fifth, there are no biologic markers for CIP. Finally, there is a generalized lack of awareness of this disorder. Thus, the diagnosis of CIP requires an awareness of the disorder combined with a carefully performed history and physical examination in addition to tests to exclude mechanical obstruction and, frequently, tests to assess gastrointestinal transit and the neuromuscular function.
Symptoms
The most common symptoms occurring in CIP are abdominal pain (80 %), nausea and vomiting (75 %), constipation (40 %), and diarrhea (20 %). The clinical picture tends to be dominated by abdominal pain and distension which are particularly severe during episodes of exacerbation. In CIP, symptoms should be present for a minimum of 6 months.
Imaging
Given the nonspecific nature of the symptoms of CIP, the initial evaluation centers on excluding mechanical obstruction. Evidence of obstruction essentially excludes the diagnosis of CIP. Although a plain X-ray of the abdomen may have findings suggestive of obstruction, cross-sectional abdominal imaging such as CT or MR is necessary to more thoroughly evaluate for mechanical obstruction. The use of barium contrast small bowel studies has largely been superseded by the development of CT and MR enterography protocols.
Endoscopy
Upper endoscopy, colonoscopy, and, occasionally, enteroscopy are useful in suspected CIP to identify intraluminal lesions, collect biopsy and fluid samples (e.g., to identify small bowel bacterial overgrowth), and, on occasion, provide treatment (e.g., placement of decompression tubes).
Motility Testing
For patients with persistent unexplained symptoms or to confirm the diagnosis of CIP, specialized tests to assess gastrointestinal motility and transit may prove useful. If symptoms of early satiety, nausea, and vomiting are predominant, a 4-h solid-phase gastric emptying scan should be performed to document the extent of delayed emptying. Similarly, colon transit testing, using either radio-opaque markers or scintigraphy, may provide useful information. The utility of small bowel transit testing remains poorly understood, and, when determined by lactulose hydrogen breath testing, accurate interpretation may be limited by the presence of small bowel bacterial overgrowth.
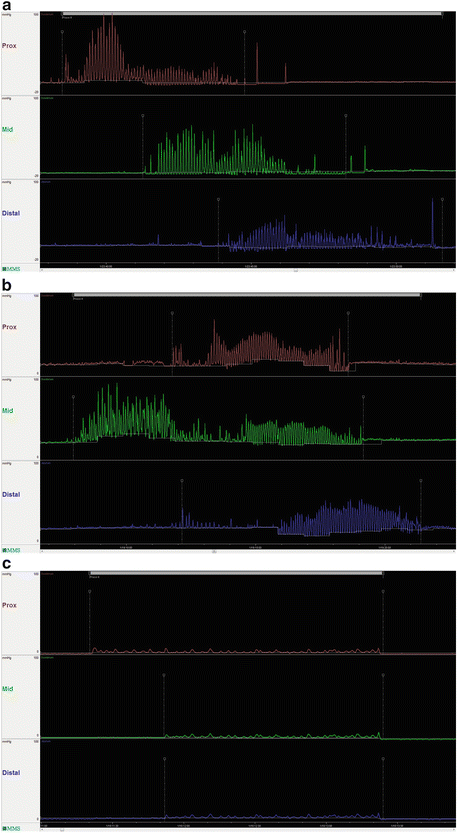
Further support for the diagnosis of CIP, and clues to the possible underlying etiology, may be obtained using intraluminal GI pressure recordings (i.e., manometry). Although esophageal manometry may demonstrate findings characteristic of scleroderma-related CIP, it mostly reveals nonspecific abnormalities. Indeed, nonspecific findings in esophageal motility have been found in more than 70 % of patients with CIP. Esophageal manometry may prove useful in predicting who will need parenteral nutrition as one study has shown that patients with ineffective esophageal motility were more likely to require parenteral nutrition than those with normal motility. Although not widely available, antroduodenal manometry (and small bowel manometry) may reveal characteristic neuropathic and myopathic abnormalities of the migrating motor complex (MMC) during the fasting and fed periods and may help to differentiate mechanical obstruction from CIP. Myopathic (smooth muscle) disorders are characterized by abnormally low-amplitude, coordinated contractions, whereas neuropathic processes are characterized by uncoordinated contractions and the absence of an MMC (Fig. 12.2). The presence of an MMC on antroduodenal manometry has been suggested to be predictive of successful tolerance to jejunal feeding in patients who have previously failed gastrostomy feeding. During antroduodenal manometry, patients with suspected CIP may be challenged with erythromycin (to stimulate gastric contractions) and octreotide (to stimulate small bowel motility); however, the clinical utility of these drug challenges remains to be demonstrated. Wireless motility capsule testing, the most recent addition to the motility testing armamentarium, transmits intraluminal pH, temperature, and pressure data allowing a determination of gastric, small bowel, and colon transit times. The role of this test in the evaluation of CIP, however, has not yet been established.
 < div class='tao-gold-member'>
< div class='tao-gold-member'>




Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
