Fig. 7.1
Mild hepatocanalicular cholestasis (arrow) in a patient with acute Hepatitis A virus infection. Note the background lobular disarray and inflammation. H and E, 600×. (Courtesy of Maxwell L. Smith, MD)
The precise mechanism of HAV cholestasis is unknown. Cholestasis by viral infection may be induced by inhibition of bile salt transporter function by proinflammatory cytokines, specifically tumor necrosis factor-alpha (TNF-a) and interleukin-1 (IL-1). Multidrug resistance-associated protein 2 (mrp2) is a transporter protein that is involved in the excretion of bilirubin; this protein is inhibited by TNF-a and IL-1 [11]. It has not been elucidated if virologic or host attributes are the primary factor. Jung et al. postulate that features of the host have a more significant role [6]. A study by Krawczyk et al. investigated genetic polymorphisms of different protein transporters responsible for bile secretion with certain genotypes associated with cholestasis—ABCB4 and ABCB11 [12]. The prevalence of the ABCB11 variant reaches 20–50 % in Europe; this potentially at-risk group may benefit from HAV vaccination. In contrast, Coppola et al. state that certain genotypes of HAV (Ia and Ib) may account for cholestasis and subsequently a more severe course of illness [13]. Further studies are required to better define the etiology of this disease process.
Cholestasis will spontaneously resolve and the prognosis is good, though the majority of patients have a protracted course. This may result in extended length of hospital stay with associated increase in medical expenses. The use of corticosteroids has been discussed in the literature. It will hasten resolution of pruritus and result an improvement in bilirubin in two-thirds of patients [14]. However, there is a concern that steroids may predispose patients to relapse [15]. If pursued, the standard regimen is prednisone 40 mg po daily, then gradually tapered over 4 weeks. Other investigational treatments include ursodeoxycholic acid [16], rifampin and plasmapheresis [12].
Hepatitis B
Cholestatic viral hepatitis was first described in patients with hepatitis B by Davies et al. [17]. The most severe and ominous form is a distinct clinical and histologic entity known as fibrosing cholestatic hepatitis (FCH). The clinical course is characterized by progressive jaundice, mildly elevated aminotransferase levels, high levels of viremia, prolongation of prothrombin time, and rapid progression to liver failure evidenced by encephalopathy. It is primarily seen in recipients of liver allografts. However, it has also been identified overall in the setting of immunosuppression such as patients following renal transplantation [18–21], bone marrow transplantation [22], patients with HIV [23], and autoimmune diseases requiring high dose immunosuppression [24].
Histologically, there is ballooning degeneration of hepatocytes with ground-glass cytoplasm, and extensive periportal fibrosis. There is also severe canalicular and intracellular cholestasis. Interestingly, there are only mild inflammatory infiltrates. Notably, hepatitis B surface antigen and hepatitis B core antigen expression are prominent in the hepatocytes. As a result, the pathogenesis does not appear to be mediated by a host immune response, as typically seen in the immunocompetent patient. Instead, in the setting of immunosuppression the replication of the hepatitis B virus appears to have a direct cytopathic effect. Accumulation of hepatitis B surface antigen and hepatitis B core antigen in hepatocytes in the endoplasmic reticulum damages vital cell functions, leading to cell death [25].
Prior to the initiation of hepatitis B immunoglobulin (HBIG) and effective oral antiviral agents, recurrence of hepatitis B post-transplant was a frequent occurrence with the risk of development of FCH increased among those with the pre-core variant [26]. Pre-core mutant strains are able to undergo unrelenting viral replication because the lack of the hepatitis B e antigen enables the virus to avoid immune recognition. The prognosis of FCH is poor, with graft loss, liver failure, and death within weeks to months of onset. As a result, in the early 1990s, many transplant centers elected to deny transplantation to patients with chronic HBV. Initially, antiviral agents were used for the treatment of FCH including acyclovir, zidovudine, and ganciclovir. However, the advent of nucleoside and nucleotide analogues resulted in significant improvement in the outcomes of post-transplant HBV [27]. Due to the fact that liver injury in FCH is secondary to direct toxicity of high levels of HBV DNA, the majority of the literature has evaluated the role of lamivudine for treatment [28, 29]. The mechanism of action of a nucleoside analogue is inhibition of DNA synthesis by terminating the nascent proviral DNA chain through interference with the reverse transcriptase activity of HBV [29]. The standard dose of lamivudine is 100 mg administered daily, with suppression of HBV DNA to undetectable levels in 96 % of patients with chronic hepatitis B [30]. It can be given either alone or with HBIG. It has been shown to rapidly reduce the levels of HBV DNA, with subsequent improvement in liver function in a few months. However, the emergence of lamivudine resistance with prolonged use leading to the development of viral mutants is a notable concern. This has been observed in up to 14 % of patients after 1 year of therapy. Two mutations at codon 552 with substitution of methionine by isoleucine (M552I) or valine (M552V) have been identified [31]. As a result, newer agents such as tenofovir and entecavir have been used with promising results as they have in vitro as well as in vivo activities against lamivudine-resistant mutants [32]. For those patients who are still viremic at time of transplant, treatment with tenofovir or entecavir should be pursued to minimize the risk of developing FCH. Immunosuppression with tacrolimus rather than cyclosporine is favored [34]. A steroid sparing regimen is preferred, but if prednisone is required, it should be tapered quickly as it may potentiate HBV replication via the glucocorticoid-responsive element on the HBV genome [33]. If the above measures are ineffective, retransplantation may be required, with the caveat that FCH may recur [17, 23, 34].
Hepatitis C
Cholestatic hepatitis C primarily follows liver transplantation. Recurrent hepatitis C in the transplanted liver is nearly inevitable; with 70–90 % having histologic evidence of recurrence and up to 20 % of recipients developing cirrhosis within 5 years of transplantation. An infrequent but aggressive form of recurrence is cholestatic hepatitis C. It primarily affects patients who are immunosuppressed due to cytotoxic therapy, human immunodeficiency virus, bone marrow transplantation, or solid organ transplantation of the kidney, heart, or liver. Jaundice, high alkaline phosphatase and gamma-glutamyl transpeptidase levels, and very high serum HCV RNA levels characterize it. Cholestatic HCV has been reported to affect between 2 and 15 % of hepatitis C infected liver transplant recipients [35, 36]. The International Liver Transplant Society defined this entity in 2003 by the following criteria: occurs more than 1 month post-transplantation, patient is significantly immunosuppressed, serum bilirubin is great that 6 mg/dl, serum alkaline phosphatase and GTT are greater than five times the upper limit of normal, hepatitis C viral load is very high, histologic features of hepatocyte ballooning in the perivenular zone, absence of hepatic artery thrombosis and biliary strictures [37].
The pathogenesis of cholestatic HCV is hypothesized to be a consequence of direct viral cytotoxicity in the setting of immunosuppression (Fig. 7.2). The medications required to prevent rejection (i.e., corticosteroids, tacrolimus, mycophenolate mofetil) lead to increased HCV entry into hepatocytes via upregulation of the entry receptors occludin and scavenger receptor B1 (SRB1). However, the virus does not follow the same kinetics in all patients, and hence the variable clinical outcome of recurrent hepatitis C. In cholestatic HCV, there is a predominately Th2 cytokine response (IL-4/Il-10) at the messenger RNA and protein level. Conversely, in non-cholestatic recurrent HCV, the response is primarily Th1 with expression of IL-2 and IFN-gamma. Other data have investigated genetic diversification and quasispecies. A quasispecies is a population of closely related yet unique genomic RNA viral sequences; in cholestatic HCV the quasispecies become more homogenous [35]. The combination of these factors leads to high viral replication. Within the hepatocyte nucleus, the virus activates cell death, induces Il-8, and expresses high levels of interferon-stimulated genes. Histologic changes then become evident with hepatic inflammation, hepatocyte ballooning, and cholestasis, ultimately leading to allograft failure [38].
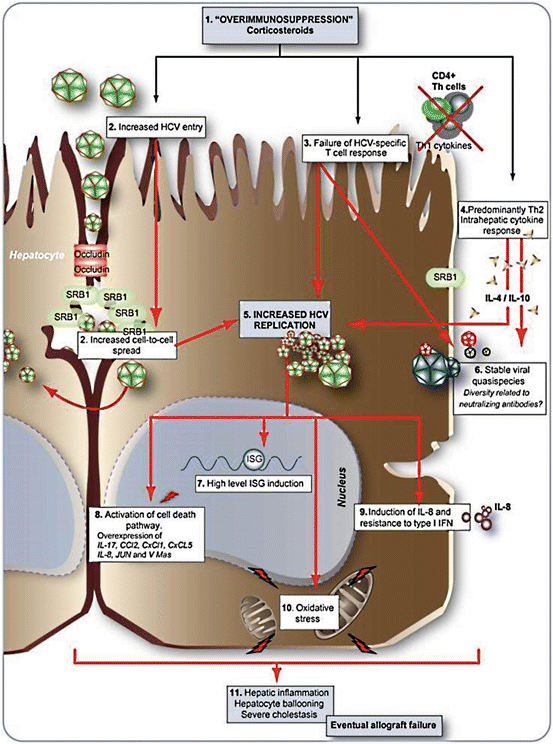
Fig. 7.2
Pathogenesis of cholestatic hepatitis C post liver transplantation. From McCaughan GW, Bowen DG. Pathogenesis of cholestatic hepatitis C. J Hepatol;54(2):392–4. Reprinted with permission from Elsevier Limited
Early recognition is imperative as the disease has a rapid progression to liver failure and death independent of cirrhosis or fibrosis. A recent study by Verna et al. identified clinical and histologic features among patients with cholestatic HCV. Higher MELD scores at transplantation, older donor age, higher incidence of biopsy-proven acute cellular rejection (Banff grade >5), and total bilirubin >2 mg/dl were all predictive of cholestatic HCV. Interestingly, HCV viral load was not predictive of cholestatic HCV in this study [36]. Hanoumeh et al. found that donor IL28B genotype TT was associated with a higher incidence of cholestatic HCV. There was no association with the recipient IL28 receptor polymorphism [39]. On liver biopsy, patients with three out of the four following features had an increased risk of cholestatic HCV: prominent ductular reaction resembling a biliary obstruction, canalicular bile plugging and/or intracellular bile pigment, prominent hepatocyte ballooning with lobular disarray, and any degree of fibrosis (especially sinusoidal) (Fig. 7.3).
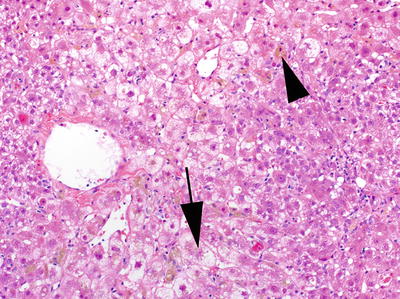
Fig. 7.3
Marked cholestasis (arrow head) with ballooning degeneration (arrow) in a centrilobular distribution in a patient with post-liver transplant fibrosing cholestatic Hepatitis C virus infection. H&E, 200×. (Courtesy of Maxwell L. Smith, MD)
Distinguishing cholestatic HCV from other forms of chronic hepatitis in liver allografts is usually difficult and requires correlation with clinical, biochemical, and serologic profiles. Clinical risk factors include genotype (specifically 1b), immunosuppression, and high HCV viral load. Unfortunately, the available literature does not regularly report these details to confirm their relevance. In cholestatic HCV, the earliest histologic features are generally lobular without significant portal changes. This is distinct from acute cellular rejection, which typically displays endothelialitis, cholangitis, and mixed inflammatory cellular infiltrates. Clinically, patients exhibiting acute cellular rejection usually have an adjustment in the immune suppression protocol as the inciting event. Non-cholestatic recurrent hepatitis C shows chronic portal and lobular inflammation and acidophil bodies with or without fibrosis.
Graft loss has been reported to occur within 1–2 years after transplantation among patients with cholestatic HCV. Treatment of those patients is complex and should be initiated promptly to help salvage the liver transplant and prevent graft failure. The immunosuppression regimen should be modified and minimized. This will include a rapid steroid taper, elimination of mycophenolate mofetil, and in some patients conversion from tacrolimus to cyclosporine, as cyclosporine may have antiviral properties and inhibit HCV replication. Antiviral therapy should be initiated as well if patient clinical status will allow. Most published data is in the form of case series, evaluating the efficacy of pegylated interferon and ribavirin. In one of the largest case series, Aqel et al. described the experience of 17 patients with FCH. Fourteen patients qualified for treatment and viral clearance was only achieved in three patients—18 % of the entire cohort [40]. In another case series by Gopal and Rosen, seven patients with cholestasis were treated with interferon alfa-2b with ribavirin. Four had a response evidenced by an undetectable viral load and normalization of bilirubin. Two patients discontinued therapy with a rebound in cholestasis. The only two patients who survived required therapy indefinitely [41]. To date, there has yet to be any published data on the use or efficacy of the newer agents for HCV (protease and/or polymerase inhibitors) for the treatment of post-transplant cholestatic HCV. Several ongoing trials with interferon-free regimens are showing promising results for recurrent HCV post-transplant including patients with cholestatic HCV.
The prognosis of cholestatic HCV remains poor with nearly 50 % mortality even after treatment with antivirals. In a systematic review by Narang et al., 47 of 94 patients died between 2 months to 312 days after transplantation. Cholestatic HCV is considered a contraindication for retransplantation in most transplant centers given poor outcomes observed.
Hepatitis E
Hepatitis E (HEV) is a small spherical virus belonging to the genus Hepevirus. It is a linear, single-stranded, positive sense RNA virus. Hepatitis E is an enterically transmitted or food-borne disease with reservoirs in humans, swine, boar, and deer [42]. This distinct hepatotropic virus was first recognized in 1980 as an epidemic in Kashmir, India [43]. Other endemic areas include Pakistan and China [42]. It is uncommon in the USA or other developed countries.
HEV accounts for more than 50 % of acute viral hepatitis in young adults in developing nations. Clinically, HEV causes a self-limited illness with moderate jaundice and normalization of aminotransferases within 1–6 weeks [44]. Risk factors for hepatitis E infection include older age (median age 53 years) and recent travel history. Hyperbilirubinemia, coagulopathy, and hypoalbuminemia are more severe compared to hepatitis A [42]. There are cases of fatality (0.5–3 %); this is particularly among pregnant women with mortality up to 25 % [45]. No chronic sequelae have been identified.
Approximately 25 % of patients develop cholestatic features with acute HEV, evidenced by prolonged jaundice, pruritus, and acholic stools [46]. Compared to hepatitis A, protracted cholestasis is more prevalent with hepatitis E infection. Histopathology shows cholestasis with bile plugs in the canaliculi, feathery degeneration of hepatocytes and liver cell rosettes. In addition, lobular inflammation and fatty changes of the hepatocytes may be seen [45]. Treatment is supportive care.
Cytomegalovirus
Cytomegalovirus (CMV) is a herpes non-hepatotropic virus. Primary infection by CMV generally occurs subclinically as a mild flu-like syndrome with subsequent life-long latency within the host. Infection closely resembles EBV infectious mononucleosis with fever, atypical lymphocytosis, and splenomegaly. Liver involvement is manifested as subtle elevations in the transaminases. It is primarily seen among transplant recipients or in patients with other states of immunosuppression [47], though it can occasionally infect the immunocompetent patient as well [48].
The cholestatic variant is quite rare and only mentioned in a few case reports in the literature [49–53]. Diagnosis may be supported by an elevation in CMV PCR; however a liver biopsy is often necessary. Histopathology illustrates cholestasis in the form of canalicular biliary thrombi and an irregularly distributed lymphocytic infiltrate. With immunohistochemistry, CMV intranuclear inclusions are present. Extrahepatic biliary obstruction and/or dilation is also associated with CMV, and therefore an MRCP and/or ERCP may be pursued [47].
CMV cholestatic liver disease will spontaneously resolve with supportive care. However, there are cases of fatality and severe disease can be treated with ganciclovir [50].
Epstein-Barr Virus
EBV is a non-hepatotropic virus; it belongs to the human herpesvirus family [54]. More than 90 % of healthy subjects chronically carry EBV in B-lymphocytes. It is transmitted through close personal contact (via saliva, genital secretions) among children and adolescents. Infection classically causes a syndrome of fever, an exudative pharyngitis, fatigue, splenomegaly, and lymphadenopathy, known as infectious mononucleosis. Atypical lymphocytosis is a common biochemical feature. Liver involvement is frequent, affecting 90 % of patients. It is usually manifested by a mild and transitory elevation in transaminases.
This disease can present primarily with cholestasis, with one study reporting 65 % among all patients infected with EBV [55]. Jaundice occurs in only 6.6 % of patients [56]. Diagnosis is made by a positive EBV IgM antibody. Abdominal ultrasound usually illustrates hepatosplenomegaly, however there is occasional gallbladder wall thickening [56, 57]. Although not necessary, if a liver biopsy is pursued, inflammatory changes are noted with apoptotic hepatocytes, a mononuclear lymphocytic infiltration, granulomas, and centrilobular cholestasis.
Treatment is largely supportive. Steroids and antivirals (i.e., acyclovir, ganciclovir) have been used to treat severe cases; however randomized control trials have never been pursued. Some studies suggest use of ursodeoxycholic acid or cholestyramine to treat pruritic symptoms related to cholestasis. Molecular absorbent recycling system (MARS) has also been employed for severe cholestasis with subsequent decrease in hepatic encephalopathy [56]. Prognosis is favorable with natural recovery in weeks without concern for chronic liver disease.
The mechanism of EBV-induced cholestasis is not known. Theories have postulated that the virus inhibits MRP2, the main bilirubin transporter [56]. Other studies suggest that cytokines interfere with the sinusoidal and canalicular transporting systems that may lead to cholestasis [55] (Table 7.1).
Table 7.1
Cholestasis in viral disease
Unique laboratory abnormalities | Histology | Course | Treatment | |
|---|---|---|---|---|
Hepatotropic viruses | ||||
Hepatitis A | • IgM HAV | • Centrilobular cholestasis • Portal inflammation | Self-limiting | • Supportive • Steroids if severe and prolonged disease • UDCA |
Hepatitis B | • Elevated HBV DNA | • Perisinusoidal fibrosis • Cholestasis • Ground-glass transformation • Hepatocyte ballooning • Mild inflammatory reaction | Rapidly progressive graft failure | • Lamivudine +/− HBIG • Adefovir • Reduce immunosuppression • Retransplanta-tion |
Hepatitis C | • Elevated HCV RNA • Genotype 1b | • Perisinusoidal fibrosis
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||



