Canalicular impairment
Sepsis
Hormone mediated cholestasis (intrahepatic cholestasis of pregnancy)
Congestive hepatopathy
Paraneoplastic cholestasis
Systemic amyloidosis
Sickle cell disease
Small intrahepatic bile ducts obstruction
Sarcoidosis
Rheumatological diseases
Cystic fibrosis
Graft versus host disease
Extrahepatic malignancies
Large intrahepatic bile duct obstruction
HIV cholangiopathy
Cholestasis Due to Impaired Canalicular Function
Sepsis
After malignant biliary obstruction, extrahepatic infections and sepsis-associated cholestasis are the second most common cause of jaundice in hospitalized patients [1]. Cholestasis has been reported in 10–20 % of patients with sepsis; both gram-positive and gram-negative organisms are identified as etiologic agents [1, 2]. Cholestasis may occur early in the course of systemic infection, even before documented bacteriological diagnosis is made, or may not appear until clinical signs of active infection have resolved [3]. Sepsis-associated cholestasis is associated predominantly with jaundice (conjugated bilirubin level usually 5–10 mg/dL), in the setting of normal or moderately elevated alkaline phosphatase and aminotransferases. The mechanisms of cholestasis in sepsis have been studied in experimental models. Circulating bacterial lipopolysaccharides (LPS) induce Kupffer cells to release excessive pro-inflammatory cytokines including tumor necrosis factor (TNF), interleukin-1β, and interleukin-6 (IL-6) into the bloodstream [4]. These proinflammatory cytokines are potent inhibitors of hepatocellular transport mechanisms and cause down regulation of key bile acid transporters such as sodium taurocholate cotransporting polypeptide (NTCP), multidrug resistance-associated protein 2 (MRP2) and bile salt export pump (BSEP) resulting in the impairment of bile flow [5–7]. Sepsis is also associated with an increased release of nitric oxide by Kupffer and endothelial cells, which in turn disrupts bile canalicular transportation and ductular bile secretion resulting in cholestasis [8, 9].
Diagnosis of sepsis-induced cholestasis can be clinically challenging due to the frequent presence of confounding comorbidities including acalculous cholecystitis, extrahepatic biliary obstruction and drug induced liver injury. Liver biopsy is not routinely recommended and when obtained reveals bland intrahepatic cholestasis without associated inflammation (Fig. 8.1) [10]. The presence of massive dilatation of bile ductules at the margin of portal tracts with inspissated bile (cholangitis lenta) is seen in patients with indolent sepsis and is associated with poor prognosis [11, 12]. Management of sepsis-related cholestasis focuses on the treatment of underlying infection and hemodynamic instability with appropriate antimicrobial therapy and aggressive supportive care. Additionally, corticosteroids may be considered as possible therapeutic options in the treatment of septic shock. Sepsis induced inflammatory cytokines such as TNF-alpha and IL-6 decrease cortisol production from the adrenal gland, inducing corticosteroid resistance and relative adrenal insufficiency [13]. Low-dose steroid therapy is associated with improved blood pressure and duration of vasopressor support but not mortality in patients with septic shock [14, 15].
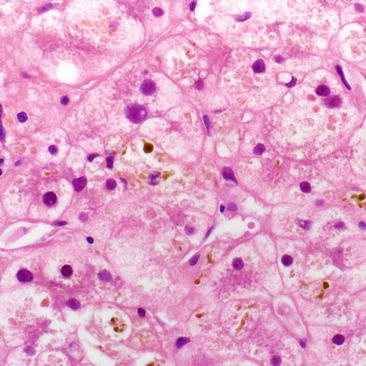
Fig. 8.1
Hepatic lobule showing severe cholestasis within hepatocyte cytoplasm and within bile canaliculi. There is associated ballooning degeneration of hepatocytes secondary to the emulsifier effects of bile. No significant lobular inflammation is in the background. This is the typical appearance of any cholestatic disease, including drug, infection, sepsis, intrahepatic cholestasis of pregnancy, etc. (H&E, 400×)
Hormone Mediated Cholestasis
Estrogen related cholestasis is well known to occur in the setting of oral contraceptive use and in intrahepatic cholestasis of pregnancy (ICP) in susceptible women. In animal models, estrogen glucuronides inhibit canalicular bile secretory processes via interactions with proteins such as BSEP and MRP2 [16, 17]. Estrogens also alter the expression of liver sinusoidal transporters and reduce bile flow [18]. Progesterone and its metabolites can trigger cholestasis by inhibiting bile acid uptake and canalicular transporters as demonstrated in animal studies [19, 20]. Progesterone and its metabolites increase during pregnancy and elevate further in ICP, potentially contributing to the pathogenesis of cholestasis.
ICP, the most common liver disease associated with pregnancy, is characterized by pruritus, elevated bilirubin and alkaline phosphatase and higher serum bile acid concentration. The symptoms usually begin in the second or third trimester of pregnancy and resolve spontaneously within 2–3 weeks after delivery. The incidence of ICP varies from 0.05 to 20 % of pregnancies and genetic, hormonal, and environmental factors may contribute to the pathogenesis [21]. Familial clustering, a higher incidence in certain ethnic groups, and geographic distribution in specific regions of Chile, Bolivia, and Scandinavia support a genetic predisposition for ICP. Heterozygous mutations in the canalicular phospholipid translocator ATP-binding cassette 4/multidrug resistance 3 (ABCB4/MDR3) involved in phosphatidylcholine excretion into the bile have been demonstrated in up to 20 % of women with ICP [22–24]. Mutations in another canalicular transporter, BSEP (ATP-binding cassette, subfamily B member 11 (ABC11) gene) which is critically involved in secretion of bile salts from hepatocytes into the bile, has also been associated with the development of ICP [25, 26]. Additionally, genetic variations in nuclear farnesoid X receptor (FXR), which plays a key role in maintaining bile acid homeostasis by regulating genes involved in bile acid metabolism and transport, have been linked with ICP [27]. Finally, cholestasis can result from estrogen and progesterone metabolites impairing hepatic transporters (BSEP), especially in genetically predisposed women [16, 20].
Pruritus is the predominant symptom in ICP and jaundice is reported in 10–25 % of patients [28]. Fasting serum bile acid concentration greater than 10 mmol/L is a sensitive diagnostic marker for ICP, although this test is not often clinically available. Liver biopsy is not necessary for diagnosis and if performed will show cholestasis and bile plugs predominantly in zone 3 [29]. In 45–70 % of women, ICP recurs in subsequent pregnancies [30]. Maternal prognosis is favorable, but affected women remain at increased lifetime risk of gallstone formation [31]. ICP poses risk for the fetus with an increased risk of premature delivery (60 %) and death [27].
Ursodeoxycholic acid (UDCA, 10–15 mg/kg) is considered the treatment of choice for ICP. UDCA therapy has been shown to reduce pruritus, improve liver test abnormalities [32–35] and also benefit fetal outcomes [36, 37]. Other pharmacologic agents such as cholestyramine, dexamethasone, and S-adenosyl-methionine were found to be ineffective compared to UDCA in patients with ICP [35].
Cholestasis Due to Cardiac Disease
Congestive heart failure, especially right-sided heart failure, is associated with a cholestatic pattern of liver injury [38–41]. Laboratory studies reveal mild to moderate elevation of total bilirubin, moderate increase in alkaline phosphatase and γ-glutamyl transferase with mild alterations in aminotransferases [36]. Prolonged prothrombin time, reduced serum albumin, right upper quadrant pain and ascites can also be seen in some patients with chronic congestive heart failure, making it difficult to distinguish between passive congestion and more advanced liver disease with synthetic dysfunction [37]. Microscopic examination of liver demonstrates sinusoidal dilatation, hemorrhagic necrosis in zone 3, bile thrombi with varying degree of cholestasis, and centrilobular and periportal fibrosis with eventual progression to bridging fibrosis (cardiac sclerosis) [35, 42].
The contributing factors for cholestasis in chronic heart failure are poorly understood. Hemodynamic data suggest that the elevated right atrial pressure and central venous pressure result in backward congestion of hepatic venules and sinusoids leading to hepatocellular dysfunction. Chronic passive congestion triggers sinusoidal collagen formation and fibrosis in the perivenular area leading to disruption of blood flow and hepatocyte atrophy [35, 36]. Animal data suggest that the increase in hepatic intrasinusoidal pressure may potentially disrupt the intrahepatocyte tight junctions (zonula occludens) creating communication between the canaliculus and the sinusoid allowing the bile contents back into systemic circulation [43].
Recent studies demonstrate that the degree of cholestasis is related to the severity of heart failure as assessed by the presence of tricuspid regurgitation and increase in right atrial pressure [39, 41, 44–48]. Cholestasis in heart failure has prognostic significance, as elevated alkaline phosphatase or bilirubin has been found to be an independent predictor of all-cause mortality, cardiovascular death, and hospitalizations in patients with chronic heart failure [40, 41].
Paraneoplastic Cholestasis
Paraneoplastic cholestasis in a patient with renal cell carcinoma (Stauffer’s syndrome) was first described in 1961 by M.H Stauffer [49]. The overall incidence of paraneoplastic cholestasis in renal cell carcinoma is estimated at 15 % [50]. Stauffer syndrome is characterized by elevated alkaline phosphatase and γ-glutamyl transferase, with or without jaundice and pruritus in the absence of biliary obstruction or hepatic metastasis [51, 52]. Prolongation of prothrombin time secondary to low grade disseminated intravascular coagulation, thrombocytosis, elevated erythrocyte sedimentation rate, and hepatosplenomegaly are also associated with this syndrome [53]. Histopathology of liver demonstrates prominent sinusoidal dilatation focally in the mid-zone of the hepatic lobules and Kupffer cell hyperplasia with no evidence of hepatocellular necrosis [54, 55]. The pathogenesis of paraneoplastic cholestasis is not fully understood. Inflammatory cytokines such as IL-6 [56, 57], interleukin-1β [58], and other mediators [50, 55] have been implicated. In patients with renal cell carcinoma, serum alkaline phosphatase significantly decreased during anti-IL6 administration, indicating a role of IL-6 in the pathogenesis of Stauffer’s syndrome [56].
Appropriate treatment of the primary tumor can lead to complete resolution of cholestasis in Stauffer’s syndrome [51, 55, 59–62]. Although paraneoplastic cholestasis is associated primarily with renal cell carcinoma, it has also been described in prostate cancer [62–64], pheochromocytoma [58], medullary thyroid cancer [65], systemic mastocytosis [66, 67] soft tissue sarcoma [68], and lymphoproliferative disorders [69].
Cholestasis Resulting from Obstruction of Small Intrahepatic Ducts
Hepatic Sarcoidosis
Sarcoidosis is a multiorgan granulomatous disease characterized by the presence of noncaseating epithelioid granulomas in the affected organs. Sarcoidosis can affect any age, gender or ethnicity but is more common in young African Americans aged 30–39 years [70, 71].
The etiology of sarcoidosis is uncertain, but environmental agents, microbial triggers, and genetic associations have been implicated in the pathogenesis [72–74]. Hepatic granulomas form as a response to unknown antigenic stimulus in a predisposed host [75]. Activation of CD4 helper T lymphocytes with release of inflammatory cytokines such as IL-2, interferon gamma, interleukin 12, and TNF promote macrophage accumulation and activation resulting in initiation and maintenance of granulomas [76–78]. In response to various proinflammatory cytokines, epithelioid histiocytes, fibroblasts, and lymphocytes accumulate and form clusters resulting in granulomas [79]. De novo sarcoidosis has been reported during and following treatment with pegylated interferon alfa 2b and ribavirin, suggesting the immunomodulatory role of interferon alfa in the initiation of sarcoidosis [80–82].
The diagnosis of hepatic sarcoidosis is based on clinical presentation, biochemical parameters and histology. Most patients with hepatic sarcoid are asymptomatic [83, 84] and only a minority develop cholestasis, portal hypertension, Budd–Chiari syndrome, or cirrhosis [85–90]. Laboratory profile demonstrates elevated alkaline phosphatase [91], moderately elevated total bilirubin, mild increase in aminotransferase levels, and mild hypergammaglobulinemia [92]. Histology reveals noncaseating granulomas found preferentially within the portal triads although they can occasionally be seen in the liver lobules. Accumulation of granulomas in the portal tracts causes destruction of the septal and interlobular bile ducts, eventually leading to chronic cholestasis, ductopenia, and pathology similar to primary biliary cirrhosis [75, 87, 93]. Rarely, sarcoid granulomas affect large bile ducts resulting in severe cholestasis and cholangiographic features similar to primary sclerosing cholangitis [94–97]. In advanced cases, severe ductopenia, dense periportal fibrosis, or cirrhosis can occur [85].
No consensus exists regarding the treatment of hepatic sarcoidosis. Asymptomatic patients with biochemical abnormalities can be observed closely without treatment [98]. Conversely, corticosteroids may be considered in symptomatic patients with hepatic dysfunction [99, 100]. Steroid therapy may result in improvement in liver test abnormalities [101, 102] but does not alter the natural course or progression of disease [103, 104]. Despite steroid therapy, progression of portal hypertension and fibrosis has been noted [88]. Other nonsteroidal therapies reported to have anecdotal efficacy include UDCA [105, 106], methotrexate [107], infliximab [108, 109], and chloroquine [98]. Liver transplantation in advanced sarcoidosis results in good patient and graft outcome [110]. However, the United Network for Organ Sharing/Organ Procurement and Transplantation Network (UNOS/OPTN) analysis showed a slightly worse allograft and patient survival compared to transplantation for other cholestatic liver diseases [111]. Although post-transplant immunosuppression may be helpful to suppress granuloma formation, recurrent disease in the transplanted organ may occur [110–112].
Connective Tissue Diseases
Abnormal liver tests in rheumatologic disorders are highly variable, ranging from asymptomatic elevated liver function tests to cirrhosis [113, 114]. Elevated alkaline phosphatase has been reported in up to 25–50 % of patients with systemic lupus erythematosus [115–117], 18–50 % of rheumatoid arthritis [118, 119], up to 40 % in Felty’s syndrome [120], 27–49 % in primary Sjögren’s syndrome [121, 122], and 62 % of polymyalgia rheumatica patients [123]. Numerous histological patterns have been reported in systemic rheumatic diseases and findings include nonspecific portal inflammation, granulomas, nodular regenerative hyperplasia, steatosis, fibrosis, and cirrhosis [124]. Clinically significant cholestasis is rare in most of the connective tissue disorders; concomitant primary biliary cirrhosis develops in up to 15 % of patients [118]. Furthermore, nodular regenerative hyperplasia and primary sclerosing cholangitis are also associated with rheumatologic disorders [114, 118].
Cystic Fibrosis
Cystic fibrosis (CF) is an autosomal recessive disease characterized by mutations in the cystic fibrosis transmembrane conductance receptor (CFTR), a chloride channel transporter localized to the apical membrane of secretory and absorptive epithelial cells within the liver, biliary tree and other organs. Absent or decreased CFTR results in accumulation of inspissated biliary secretions in the intrahepatic bile ducts causing obstruction to the ductular bile flow [130]. Focal obstruction combined with injury to biliary epithelium by endogeneous bile acids results in periportal fibrosis and hepatic dysfunction [131].
Clinically apparent CF-related hepatobiliary disease develops in 15–41 % of CF patients mainly during or before puberty. Disease manifestations include neonatal cholestasis, hepatic steatosis, biliary obstruction eventually leading to biliary cirrhosis in 4.5 % of patients [130, 132]. Development of portal hypertension (7.9 %) leading to ascites and varices as late complications has also been noted in long-term CF survivors [133, 134]. The diagnosis is challenging and a combination of modalities such as clinical symptoms, liver function tests, imaging and histology should be used. There is no definitive therapy for CF-related liver disease. Treatment with high-dose UCDA has been shown to improve liver test abnormalities, histopathology and also to decrease the development of advanced liver disease with long-term use, but no effect on survival has been demonstrated [135–139]. Liver transplantation for CF-related liver disease is uncommon and the adult 5-year survival rate is less than in patients transplanted for other indications [140]. Patients without varices and preserved synthetic function can be managed medically and endoscopically without surgery [141].
Graft Versus Host Disease
Graft-versus-host disease (GVHD) is a major complication of allogenic hematopoietic stem cell transplant. The liver is frequently involved and acute or chronic cholestasis may occur. The pathophysiology of GVHD is complex and involves donor T lymphocytes reacting to host alloantigens, causing a strong cytokine response and eventually end organ damage [142, 143]. Murine GVHD models demonstrate inflammatory cell infiltration in the liver with CD8+ T cells and mononuclear phagocytes causing nonsuppurative destructive cholangitis [144].
Acute GVHD usually occurs within the first 100 days post-transplant and presents with elevation in alkaline phosphatase, gamma glutamyl transpeptidase, and conjugated bilirubin [145]. Chronic GVHD is an indolent cholestatic disease occurring in about 60 % of allograft recipients. However, chronic GVHD with marked elevation of aminotransferases resembling acute hepatitis has also been reported in the literature [146, 147]. Diagnosis of hepatic GVHD can be challenging and biopsy is usually required to confirm the diagnosis. The histologic features noted are endothelialitis, lymphocytic infiltration of the portal areas, immunomodulatory cholangitis, bile duct destruction, and portal fibrosis [148, 149]. Corticosteroids, cyclosporine, and other novel immunomodulatory drugs such as tacrolimus and mycophenolate mofetil are used in the therapy of GVHD. Treatment with UCDA has also been proven to be effective in mild cases with normalization or improvement in liver test abnormalities [150, 151].
Extrahepatic Malignancies
The liver is a common site for metastasis and malignancy. Melanoma and colon, breast, and lung cancers frequently metastasize to the liver [152]. Both Hodgkin’s disease (HD) and non-Hodgkin’s lymphoma (NHL) can cause cholestatic liver disease secondary to hepatic infiltration, obstruction of small interlobular ducts, or rarely extrahepatic biliary obstruction [153–155]. Liver involvement in HD has been reported in 11–80 % of patients and in 16–43 % in NHL cases [155, 156]. Acute liver failure due to hepatic infiltration has also been described in the literature, more commonly with NHL [157, 158]. Vanishing bile duct syndrome with destruction of small intrahepatic bile ducts is a rare and poorly understood condition seen in HD patients [159–162]. Although the prognosis is poor when vanishing bile duct syndrome occurs, successful treatment with chemotherapy for HD has been reported [160, 163].
Cholestasis from Obstruction of Large Intrahepatic Ducts
Acquired immune deficiency syndrome (AIDS) cholangiopathy is a late complication of human immunodeficiency virus (HIV) infection, commonly seen in advanced stages when CD4 counts are low. Most patients present with epigastric or right upper quadrant pain, fever, and elevated alkaline phosphatase [164]. The pathogenesis is unclear and infectious agents such as cytomegalovirus and cryptosporidium have been attributed as causative agents [165–167]. Intrahepatic or extrahepatic bile duct strictures or papillary stenosis are prominent features noted [164, 166–168]. The diagnosis is based on clinical, biochemical, and imaging evidence of biliary abnormalities. Magnetic resonance cholangiopancreatography will demonstrate biliary dilation [169]; endoscopic retrograde cholangiography is indicated to exclude papillary stenosis and for therapeutic intervention. Liver biopsy demonstrates cholestasis with portal tract edema, neutrophilic infiltration, and bile duct proliferation [170]. In patients with papillary stenosis, endoscopic sphincterotomy relieves pain, but liver enzyme elevation may persist [171]. Antimicrobial therapy for opportunistic infection has been shown to be ineffective. Highly active antiretroviral therapy may recover immune status and potentially improve cholangiographic abnormalities [172]. However, despite aggressive therapy, AIDS cholangiopathy is associated with a dismal prognosis [171, 172].
Cholestasis Due to Other Causes
Hepatic Amyloidosis
Liver involvement is common in both primary (AL) and secondary (AA) amyloidosis. Primary systemic amyloidosis is derived from immunoglobulin light chains and is associated with underlying plasma dyscrasias. Although liver involvement is common in primary amyloidosis (70 %), serious clinical manifestations are rare [173]. Common findings in hepatic amyloidosis include weight loss, abdominal pain, hepatomegaly, hyposplenism, and ascites [174–176]. Common laboratory abnormalities include elevated alkaline phosphatase (>500 U/L), jaundice, and elevated monoclonal protein in serum or urine [174, 176, 177]. Case reports of spontaneous hepatic rupture and massive bleeding have been reported in patients with hepatic amyloidosis [178–180]. Liver histopathology demonstrates amyloid deposition in the sinusoids, parenchyma, and blood vessel walls [181]. Patients with primary amyloidosis have a poor prognosis with median survival of 8–12 months [174, 182]. Treatment may include systemic chemotherapy or hematopoietic stem cell transplantation.
Secondary or AA amyloidosis is commonly seen in the setting of systemic inflammatory diseases or chronic infections. The clinical manifestations in secondary amyloidosis are similar to primary amyloidosis and the management includes treatment of underlying inflammatory disorder.
Sickle Cell Disease
Abnormal liver function tests are common in patients with sickle cell disease. Hepatobiliary complications associated with homozygous sickle cell disease include: acute sickle hepatic crisis, acute hepatic sequestration, sickle cell intrahepatic cholestasis, and extrahepatic cholestasis (cholelithiasis and choledocholithiasis) [183–185]. Acute sickle hepatic crisis occurs in 10 % of patients hospitalized for pain crisis and clinical manifestations include right upper quadrant pain, fever, jaundice, and hepatomegaly [186]. Liver function tests reveal predominantly elevated bilirubin with mild increase in aminotransferases. Sickle cell intrahepatic cholestasis is a rare and severe form of sickle cell crisis with profound conjugated hyperbilirubinemia due to marked intrasinusoidal sickling of erythrocytes and hepatic ischemia [185, 187]. Chronic intrahepatic cholestasis and cirrhosis has also been reported in patients with sickle cell disease [188, 189]. Liver biopsy in sickle cell disease demonstrates sinusoidal dilation, intrasinusoidal sickling, Kupffer cell hyperplasia, and hemosiderosis [190]. Aggressive treatment of sickle cell disease with exchange transfusion [185, 188] and liver transplantation in rare cases [191, 192] has been reported in the literature.
Total Parenteral Nutrition (TPN)
Parenteral nutrition related cholestatic liver disease is discussed in detail elsewhere. TPN induced cholestasis is a multifactorial hepatobiliary complication characterized by progressive increase in conjugated bilirubin (>2 mg/dL) and alkaline phosphatase in both adult and pediatric patients treated with hospital or home parenteral nutrition. Liver biopsy reveals intrahepatic cholestasis, steatohepatitis, varying degrees of fibrosis and cirrhosis [193]. Management includes early resumption of enteral intake, aggressive treatment of sepsis, and bacterial overgrowth. UDCA has been shown to be effective in reducing TPN related intrahepatic cholestasis [194].
Summary
Major systemic disorders can impair biliary function through diverse mechanisms causing varying degrees of cholestasis. In sepsis, Kupffer cell activation by endotoxins and bacteria initiates the cascade of inflammatory cytokines, disrupting the canalicular bile flow and resulting in cholestasis. Sepsis-related cholestasis involves treatment of the underlying infection and aggressive supportive measures. Intrahepatic cholestasis of pregnancy is a complex disorder with heterogeneous etiologies including hormonal, genetic, and environmental factors usually occurring late in pregnancy. ICP is associated with adverse fetal outcomes and UCDA is considered as the mainstay of therapy. Increased central venous pressure due to chronic heart failure results in passive hepatic congestion and cholestasis. The degree of cholestasis is related to the severity of heart failure, and elevated liver function tests have long-term prognostic implications in heart failure patients. Liver involvement in sarcoidosis ranges from asymptomatic cholestatic abnormalities to advanced disease with portal hypertension and cirrhosis. Other disorders such as cystic fibrosis and various rheumatologic conditions also cause cholestasis due to disruption of bile flow in the small intralobular bile ducts. AIDS cholangiopathy occurs in advanced HIV disease with low CD4 counts and is characterized by papillary stenosis, bile duct strictures and sclerosing cholangitis. Treatment includes endoscopic sphincterotomy, biliary stenting, antimicrobial therapy, and HAART regimen. Lastly, systemic amyloidosis and sickle cell disease also involve liver and cause varying degrees of cholestasis.
References
1.
Whitehead MW, Hainsworth I, Kingham JG. The causes of obvious jaundice in South West Wales: perceptions versus reality. Gut. 2001;48(3):409–13.PubMedCentralPubMed
2.
Fan HB, Yang DL, Chen AS, Li Z, Xu LT, Ma XJ, et al. Sepsis-associated cholestasis in adult patients: a prospective study. Am J Med Sci. 2013;346:462–6.PubMed
3.
Franson TR, Hierholzer Jr WJ, LaBrecque DR. Frequency and characteristics of hyperbilirubinemia associated with bacteremia. Rev Infect Dis. 1985;7(1):1–9.PubMed
4.
Kosters A, Karpen SJ. The role of inflammation in cholestasis: clinical and basic aspects. Semin Liver Dis. 2010;30(2):186–94.PubMedCentralPubMed
5.
Whiting JF, Green RM, Rosenbluth AB, Gollan JL. Tumor necrosis factor-alpha decreases hepatocyte bile salt uptake and mediates endotoxin-induced cholestasis. Hepatology. 1995;22(4 Pt 1):1273–8.PubMed
6.
Geier A, Fickert P, Trauner M. Mechanisms of disease: mechanisms and clinical implications of cholestasis in sepsis. Nat Clin Pract Gastroenterol Hepatol. 2006;3(10):574–85.PubMed
7.
Green RM, Beier D, Gollan JL. Regulation of hepatocyte bile salt transporters by endotoxin and inflammatory cytokines in rodents. Gastroenterology. 1996;111(1):193–8.PubMed
8.
Dufour JF, Turner TJ, Arias IM. Nitric oxide blocks bile canalicular contraction by inhibiting inositol trisphosphate-dependent calcium mobilization. Gastroenterology. 1995;108(3):841–9.PubMed
9.
Spirli C, Fabris L, Duner E, Fiorotto R, Ballardini G, Roskams T, et al. Cytokine-stimulated nitric oxide production inhibits adenylyl cyclase and cAMP-dependent secretion in cholangiocytes. Gastroenterology. 2003;124(3):737–53.PubMed
10.
Lefkowitch JH. Histological assessment of cholestasis. Clin Liver Dis. 2004;8(1):27–40. v.PubMed
11.
Lefkowitch JH. Bile ductular cholestasis: an ominous histopathologic sign related to sepsis and “cholangitis lenta”. Hum Pathol. 1982;13(1):19–24.PubMed
13.
Cooper MS, Stewart PM. Corticosteroid insufficiency in acutely ill patients. N Engl J Med. 2003;348(8):727–34.PubMed
14.
Patel GP, Balk RA. Systemic steroids in severe sepsis and septic shock. Am J Respir Crit Care Med. 2012;185(2):133–9.PubMed
15.
Annane D, Bellissant E, Bollaert PE, Briegel J, Confalonieri M, De Gaudio R, et al. Corticosteroids in the treatment of severe sepsis and septic shock in adults: a systematic review. JAMA. 2009;301(22):2362–75.PubMed
16.
Stieger B, Fattinger K, Madon J, Kullak-Ublick GA, Meier PJ. Drug- and estrogen-induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology. 2000;118(2):422–30.PubMed
17.
Huang L, Smit JW, Meijer DK, Vore M. Mrp2 is essential for estradiol-17beta(beta-d-glucuronide)-induced cholestasis in rats. Hepatology. 2000;32(1):66–72.PubMed
18.
Simon FR, Fortune J, Iwahashi M, Gartung C, Wolkoff A, Sutherland E. Ethinyl estradiol cholestasis involves alterations in expression of liver sinusoidal transporters. Am J Physiol. 1996;271(6 Pt 1):G1043–52.PubMed
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


