Primary biliary cirrhosis (PBC)
Primary sclerosing cholangitis (PSC)
Autoimmune sclerosing cholangitis
Granulomatous hepatitis
Hepatic amyloidosis
Congestive hepatopathy
Hepatic sarcoidosis
Systemic viral infections
• Hepatitis Viruses (A, B, C, D, E)
• Cytomegalovirus (CMV)
• Epstein–Barr virus (EBV)
Hemophagocytic syndrome
Extrahepatic Biliary Obstruction
Biliary obstruction is one of the more common causes of jaundice. It should be ruled out in the cholestatic hospitalized patient. Abdominal imaging and/or endoscopic diagnostic studies are generally sufficient to determine the cause. If obstruction is absent, then true hepatic cholestasis is likely.
Cholestasis of Sepsis (Cholangitis Lenta)
Pathogenesis
To understand the pathology of cholestasis of sepsis, one must understand the basics of bile processing. A primary hepatic function is the removal of organic anions (such as bilirubin), cations, and bile acids from the circulation. Hepatic bile salt and anionic steroidal compound uptake is mediated by the sinusoidal Na+/Taurocholate Co-transporting Polypeptide (NTCP), which is driven by the hepatocyte cellular sodium gradient. Organic anions such as bilirubin and cations are taken across the hepatocyte basolateral membrane in a sodium-independent manner by groups of transport proteins with overlapping specificity—Organic Cation Transporters (OCTs), Organic Anion Transporting Polypeptides (OATPs), and Organic Anion Transporters (OATs) [4, 5]. Thus, the liver plays a crucial role in bile acid metabolism and secretion [6].
Following uptake and processing, the hepatocytes then secrete the bile acids into the bile canaliculus. Bile formation is both bile acid-dependent, related to the bile salt excretion, and bile acid-independent, related to active secretion of the inorganic electrolytes and solutes [7, 8]. The bile canalicular transporters are members of the adenosine triphosphate (ATP)-binding cassette (ABC) superfamily (see Phase III Reactions, below) [9]. Canalicular secretion of bile acids is medicated by the bile salt export pump (BSEP/ABCB11), which transports bile acids across a concentration gradient and is the rate limited step in their secretion [10–15]. Secretion of the non-bile acid organic anions is medicated by the multidrug resistance-associated protein 2 (MRP2/ABCC2), formerly known as the canalicular multispecific organic anion transporter (cMOAT) [15–17]. Organic cations are secreted into the bile by the multidrug resistant P-glycoprotein 1 (MDR1/ABCB1) and MDR3 (ABCB4). These membrane transporters are regulated at the level of gene transcription, RNA translation, and posttranslationally [7, 18].
Cholestasis of sepsis (CoS) has been linked predominantly to gram-negative bacterial infections, although any significant extrahepatic systemic infection can cause it [1, 7, 19–24]. Following malignant biliary obstruction, CoS is the second most common cause of jaundice in the hospitalized patient [1, 25]. Bacterial cell walls contain lipopolysaccharides (LPS), called endotoxins, which are powerful inducers of proinflammatory cytokines, such as interleukin-1 (IL1), interleukin 6 (IL6), and tumor necrosis factor alpha (TNFα). Endotoxins and the proinflammatory cytokines they induce play a key role in the development of CoS.
Endotoxins increase intestinal permeability, leading to escape of bacterial cell wall components and endotoxin into the portal venous system [26]. This results in the accumulation of endotoxin within the liver which ultimately causes significant injury [1, 27–29]. Proinflammatory cytokines subsequently secreted by the Kupffer cells play a central role in further mediating CoS [1, 27, 30]. The cytokines may lead to expression of ligands on the sinusoidal endothelial cells, Kupffer cells, and hepatocytes, stimulating margination of neutrophils with subsequent release of superoxide radicals and enzymes. This promotes further hepatocellular injury [31–36]. Endotoxins act as powerful cholestatic agents by inhibiting both hepatocellular bile acid uptake (via NTCP and OATP) and canalicular bile secretion (via BSEP and MRP2) [31, 37–41]. Endotoxemia reduces the excretion of conjugated bile acids and bilirubin-glucuronides, and decreases bile flow [7, 41]. It severely impairs both bile acid-dependent and bile acid-independent organic anion transport at the sinusoidal and canalicular membranes [37, 42–44]. The reductions in anion transport are correlated with decreased protein and mRNA levels [38, 42, 45, 46]. Bile acid-independent biliary flow and biliary HCO3 secretion are reduced [37, 47]. Pretreatment with medications which block the LPS-mediated release of cytokines significantly attenuates the reduction in bile acid secretion and bile flow, confirming the critical role of these molecules in the development of CoS [39, 43, 44, 48, 49].
Clinical
The association of jaundice and bacterial infection was described as early as 1837 [50]. Cholestasis of sepsis, historically called cholangitis lenta, should always be considered in the differential diagnosis of any hospitalized patient who develops jaundice [25]. Cholestasis of sepsis is associated with disproportionate elevations in serum alkaline phosphatase and bilirubin compared with serum aminotransferase levels [51]. An isolated elevation in the bilirubin level may be the only abnormality seen [7]. The bilirubin elevation and cholestasis may be even more pronounced in patients with underlying hepatobiliary disease, such as sinusoidal obstruction syndrome, GVHD, or other biliary tract disorders. Bilirubin levels can approach 30 mg/dL in the septic patient in this setting [51]. In the HSCT recipient, fever and infection generally occur during the first month, when the granulocyte counts are at their nadir. Elevations in bilirubin level may even precede positive blood cultures, and a continued rise in a patient with underlying liver disease is correlated with a worse outcome [7, 51]. Treatment is directed at the underlying infection.
Liver biopsy is rarely necessary for the diagnosis of CoS. When performed in the setting of uncomplicated CoS; however, the most prominent histological finding is that of pericentral canalicular or periportal ductular cholestasis [52]. This can be associated with Kupffer cell hyperplasia, portal mononuclear inflammation, focal hepatocyte dropout, and steatosis [7, 22–24, 51]. Those who are immunosuppressed often do not show evidence of the portal inflammation seen in the immunocompetent patient [31]. Patients who are at greatest risk of dying may have a more uncommon form of CoS in which the portal and periportal bile ductules are dilated with inspissated bile [53].
Parenteral Nutrition
The use of parenteral nutrition (PN) is often critical to providing nutritional support to those who are unable to be fed enterally. Its prolonged use, however, can result in multiple complications, including metabolic bone disease, line infections, and PN-associated liver disease (PNLD), particularly cholestasis [28, 54].
Extrahepatic Complications
The most common extrahepatic complications of PN in adults include the development of biliary sludge, gallstones, and acalculous cholecystitis [28, 55–58]. Complete bowel rest decreases the production of cholecystokinin (CCK) resulting in decreased gallbladder contraction, inhibition of sphincter of Oddi relaxation, and decreased biliary flow [28, 59]. This leads to stasis and biliary sludge formation [60]. Biliary sludge has been shown to be present in all patients within 6 weeks of starting PN [56, 58]. Gallstones, which become evident after about 4 months of PN, are generally pigment stones [61]. Both calculous and acalculous cholecystitis have been reported.
Intrahepatic Complications
Many patients will develop mild to moderate liver enzyme elevations upon starting PN; however, these generally stabilize by 1–2 weeks and are not usually associated with histologic findings [62]. The liver disease associated with PN can be extremely severe. It has been assumed that the longer the duration of use of PN, the greater the risk of developing chronic liver disease; however, more recently this dogma has been questioned [63]. Chronic cholestasis has been reported in over 50 % of patients and between 15 and 40 % of patients receiving PN will develop end-stage liver disease [28, 64–66]. PNLD can lead to irreversible liver failure, with mortality rates as high as 51 % [67–69].
Multiple histologic findings are seen in the setting of PNLD. Hepatic steatosis is generally the earliest finding noted in adults and may be associated with excess glucose in the PN [28]. Most patients will have some degree of hepatic steatosis following 3 or more months of PN [55]. Over time, patients develop a pattern of injury which includes cholestasis, steatosis, portal edema and inflammation, ductular reaction and ductopenia, and portal and perivenular fibrosis [63].
The etiology of PNLD is most likely multifactorial; however, the precise cause remains unknown [54, 68, 70, 71]. Multiple factors have been proposed. PN-induced changes in bile composition may lead to biliary injury, bile sludging, bile duct obstruction, and subsequent hepatocyte injury [55, 58, 72, 73]. The use of PN with excess calories, particularly in the form of carbohydrates, leads to fatty acid synthesis and development of hepatic steatosis [28]. In fact, a direct correlation has been reported between total PN calories and fatty infiltration [74]. Hyperinsulinemia subsequently develops which stimulates glycolysis, fatty acid esterification, glycogenolysis, and gluconeogenesis and inhibits fatty acid oxidation [75–77]. Lipids may also play a role in PNLD [64]. Lipid emulsions contain phytosterols and omega-6 fatty acids which have been associated with inhibition of bile acid secretion and bile acid-stimulated bile flow [78–81]. Both serum and tissue levels of antioxidants, including glutathione, β-carotene, and vitamin E, are markedly reduced in patients on PN [28, 82, 83]. Conversely, the products of oxidative injury are increased [84]. Oxidative injury and subsequent hepatocyte apoptosis also play a role in PNLD [85]. Nutrient deficiencies, such as choline, glutamine, and taurine, may also contribute to the development of PNLD [28, 86, 87].
Management
The ideal management is to maximize enteral feeding, although this is not always possible. Multiple attempts have been made to optimize the composition of PN in an effort to prevent or decrease the associated PNLD. The most common of these changes include avoiding excess calories, decreasing amino acid content, or adding lipids, glutamine, choline, and lecithin [55]. The response has been mixed; however, and PNLD remains a significant clinical problem [63, 88]. Allowing a daily PN-free period of time of 8 h or more has also been reported to improve liver enzymes [89]. The use of ursodeoxycholic acid has been shown in small studies to improve liver enzymes [90–92]. Liver transplantation may ultimately be required if the PNLD progresses to liver failure.
Drug-Induced Cholestasis
Drugs must be considered in the differential diagnosis of cholestasis in the hospitalized patient. The liver is responsible for the selective uptake, concentration, metabolism, and excretion of the majority of drugs, toxins, and xenobiotics (“substances”) introduced into the body. Each substance has a specific disposal pathway involving one or more of the soluble or membrane-bound enzymes which process these substances. The majority of substances absorbed from the gastrointestinal tract are lipophilic and water-insoluble and they must be rendered water-soluble to be excreted.
Drug Metabolism
Exogenous substances are metabolized mainly via phase I or phase II reactions, or a combination of both (Fig. 11.1) [93–95]. The final products are then moved across the canalicular or sinusoidal membranes via excretory transporters (phase III reactions) [96]. Genetic polymorphisms exist in all of these enzymes, which can lead to either decreased or increased activity, and may predispose to xenobiotic-induced cholestasis or injury [97–105].
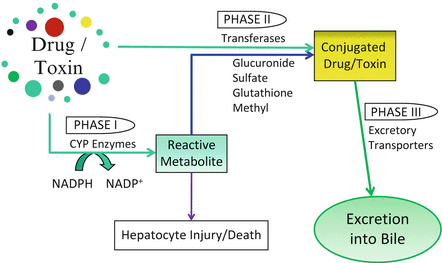
Fig. 11.1
Xenobiotic Metabolism. Exogenous substances are metabolized via phase I or phase II reactions, or a combination of both. The final products are then moved across the canalicular or sinusoidal membranes via excretory transporters (phase III reactions) for disposal
Phase I Reactions
Phase I reactions transform lipophilic molecules into more polar, hydrophilic molecules via oxidation, reduction, or hydrolysis. These reactions are catalyzed by the membrane-bound cytochrome P450 (CYP) family of mixed function oxidases [106–110]. The P450 molecules are hemoproteins composed of an apoprotein and a heme prosthetic group (the oxidizing center). There are over 50 proteins in the CYP group which are organized into 18 families (e.g., CYP2) and 43 subfamilies (e.g., CYP2E1) [111]. Most are located on the cytoplasmic side of the smooth ER membrane (microsomal-type) or the mitochondria (mitochondrial type) [111]. Phase I drug metabolism is performed predominantly via the microsomal-type. There is zonal heterogeneity of these enzymes within the liver—some are more prominent in the perivenular (zone 3) hepatocytes (i.e., P450 2E1), while others are clustered within other acinar zones. Hepatic metabolism of exogenous drugs and toxins is performed mainly by the CYP1, CYP2, and CYP3 families, with a smaller contribution from CYP4 [111–117]. The most important drug metabolizing member within the liver is CYP3A4, which comprises about 60 % of all hepatic cytochromes and catalyzes the biotransformation of over 50 % of commonly used drugs [111, 118]. Free radicals and toxic electrophilic compounds can be produced during this process.
Phase II Reactions
Phase II reactions occur either directly with a parent compound (rare) or with a metabolite formed by a phase I reaction which is still not adequately hydrophilic for excretion. These reactions conjugate the metabolic byproducts to highly polar ligands, such as glucuronate, sulfate, acetate, glycine, glutathione, or a methyl group (Fig. 11.1). The resultant water-soluble substances are nontoxic and readily excreted [95, 119, 120]. Phase II reactions occur primarily within the hepatocyte cytoplasm via the UDP-glucuronyl transferases (UGT1 and UGT2), sulfotransferases, and glutathione S-transferases [93]. Conjugation ordinarily leads to a decrease in pharmacologic activity (detoxification) and phase II enzymes are rarely responsible for toxic metabolite formation; however, exceptions do occur.
Phase III Reactions
Phase III reactions lead to the transport of compounds across the canalicular membranes and into the bile [119]. As noted above, the bile canalicular transporters include multidrug resistant P-glycoprotein 1 (MDR1/ABCB1), MDR3 (ABCB4), multidrug resistant protein 2 (MRP2/ABCC2), and the bile sale export protein (BSEP/ABCB11) [11]. Their predominant role is that of regulation of bile formation and the excretion of xenobiotics [11]. Emerging data suggests that cholestatic drug induced liver injury (DILI) is secondary to drug- or metabolite-mediated inhibition of these transporters [11, 121].
Hepatotoxicity
Because of the liver’s central role in detoxification, it is uniquely susceptible to DILI. More than a thousand drugs and herbal products have been reported to cause hepatotoxicity (Table 11.2) [122]. Those most commonly associated with cholestasis in the setting of HSCT include calcineurin inhibitors (particularly cyclosporine), azole antifungal agents, trimethoprim-sulfamethoxazole, ribavirin, busulfan, and bis-chloroethylnitrosourea [2].
Anti-infectives | • Amoxicillin–clavulanate • Erythromycin, clarithromycin, and azithromycin • Trimethoprim/sulfamethoxazole • Azole antifungal agents • Flucloxacillin, dicloxacillin • Nitrofurantoin • Isoniazid, rifampin • Ciprofloxacin • Cephalosporins • Terbinafine • HIV antivirals—stavudine, didanosine, nevirapine |
Anti-inflammatories | • Azathioprine • Diclofenac • Ibuprofen • Sulindac • Nimesulide |
Psychotropics | • Chlorpromazine • Imipramine, amitryptyline • Duloxetine • Risperidone |
Chemotherapeutics | • Busulfan • Cytarabine • Bis-chloroethylnitrosourea |
Others | • Estrogens/oral contraceptives • Anabolic steroids • Calcineurin inhibitors (i.e., cyclosporine) • ACE Inhibitors • Clopidogrel • Irbesartan • Phenothiazines |
Up to 5 % of the jaundice seen in hospitalized patients is caused by drugs [25, 123–125]. Cholestatic and mixed cholestatic/hepatocellular injury account for up to half of all drug-induced liver disease, and are its most severe presentation [121, 126–128]. Drugs dosed at over 50 mg daily are more likely to cause DILI than those with daily doses less than this amount [129, 130]. Additionally, drugs which are significantly hepatically metabolized and excreted into the bile are the most likely to produce cholestatic liver injury [131]. This injury may be manifest at any of the enzyme sites along the metabolism pathway.
Cholestasis secondary to DILI is defined as an elevated alkaline phosphatase (AlkPhos) greater than two times the upper limit of normal and/or an alanine aminotransferase (ALT) to AlkPhos ratio of less than 2 (Table 11.3) [121]. Injury is considered to be mixed if the ALT/AlkPhos ratio is greater than 2 but less than 5 and hepatocellular if this ratio is >5. The presence of jaundice (serum bilirubin >2 times the upper limit of normal) in association with an elevation in serum aminotransferases (>3 times the upper limit of normal) is associated with a worse prognosis than that seen in the setting of isolated aminotransferase abnormalities (an observation noted by Hyman Zimmerman and known as “Hy’s law”) [132, 133]. In this setting, the mortality is as high as 14 % [121, 126, 134–136].
Hepatitis (hepatocellular) | Cholestasis | Mixed |
|---|---|---|
ALT ≥3 × ULN | AlkPhos ≥2 × ULN | ALT ≥3 × ULN AlkPhos ≥2 × ULN |
R ≥ 5 | R ≤ 2 | R > 2 to <5 |
Clinical Features
DILI must remain in the differential diagnosis of all patients presenting with cholestasis. Drug-induced cholestatic liver injury can present either acutely or chronically, mimicking many extrahepatic and intrahepatic liver diseases. Presentation may be weeks to months after beginning the offending agent. Patients may be asymptomatic with only isolated elevations in alkaline phosphatase or they may be frankly jaundiced and symptomatic [122]. Fever, nausea, anorexia, malaise, fatigue, pruritus, and/or abdominal pain or discomfort may also be present. Cholestatic DILI is more common in those over 60 years of age and in those with certain genetic polymorphisms, such as HLA DRB1*15 and HLADQB1*06 [129, 137, 138]. It remains unclear whether underlying liver disease is a predisposing risk factor for cholestatic DILI [121]. Cholestasis and jaundice tend to be prolonged after discontinuation of the offending agent, and can take weeks to months to resolve. If prolonged, patients may develop xanthomas, pruritus, and melanoderma [122].
Diagnosis
The diagnosis of DILI can be difficult. Prior to attributing cholestasis to a certain drug or toxin, a complete evaluation must be undertaken to rule out other causes. Abdominal imaging may be necessary to rule out obstructive disease. Cholestasis secondary to sepsis, TPN or heart failure must be eliminated [121]. Certain infections, such as cholestatic hepatitis A virus, Epstein–Barr virus, typhoid fever, or Q fever can also present with a cholestatic picture [121, 139–141]. Obtaining a careful drug history is important but not always reliable—the relationship between exposure to the drug and cholestasis is not always clear. Hospitalized patients are often taking multiple medications, making identification of a single offending agent difficult. Extrahepatic manifestations caused by the offending agent, such as rash, interstitial nephritis, or other organ toxicity may be helpful in eliciting the diagnosis [122].
There is no gold standard, specific serum biomarker or characteristic histologic feature that reliably identifies a drug as the cause of toxicity [121]. The Council for International Organizations of Medical Sciences (CIOMS) developed a series of standard designations of drug-induced liver disorders and classification of injury [142]. The US FDA Drug Hepatotoxicity Steering Committee proposed modifications to the CIOMS scheme for classifying hepatotoxicity in clinical trials (Table 11.3) [143]. The most commonly used scale to codify causality of drug toxicity is CIOMS Roussel-Uclaf Causality Assessment Method (RUCAM) scale [142, 144, 145]. These assessments do not address all risk factors in all patients, and none are used routinely in clinical practice.
Features which suggest drug toxicity include a lack of illness prior to ingesting the drug, clinical illness or biochemical abnormalities developing after beginning the drug, and improvement after the drug is withdrawn. The illness will generally recur upon reintroduction of the offending substance if the DILI is immunologic; therefore, rechallenge is not advised. Nonspecific symptoms developing after introduction of a drug (such as nausea, anorexia, malaise, fatigue, right upper quadrant pain, or pruritus) may indicate drug toxicity and should prompt evaluation. Key elements for attributing liver injury to a drug include [146]:
Exposure must precede the onset of liver injury (although the latent period is highly variable)
Underlying liver disease should be excluded
Injury may improve when the drug is stopped (although in some cases injury may initially worsen for days or weeks, while in fulminant cases, declining liver biochemical tests may indicate deterioration rather than improvement)
Liver injury may have recurred more rapidly and severely after repeated exposure
Histologically, drug-induced cholestatic liver injury can be classified as either bland, without associated inflammation, or as cholestatic hepatitis, with inflammatory changes [147]. These changes may include portal inflammation, portal neutrophils and plasma cells, fibrosis, and intracellular cholestasis and can resemble autoimmune hepatitis [148]. DILI most often leads to cholestatic hepatitis [121, 147].
Treatment
No specific therapies have been shown to alter the clinical course of DILI. Most cases of mild cholestatic DILI will resolve with discontinuation of the drug which caused it. Chronic progressive disease can develop; however, potentially leading to vanishing bile duct syndrome, biliary cirrhosis, and ultimately progression to liver failure and death [121, 135, 149]. Patients with cholestatic DILI are more likely to develop chronic liver disease than those with hepatocellular DILI [121, 150]. Liver transplantation can be considered in appropriate patients who develop liver failure.
Lymphoma
Liver involvement with either Hodgkin or, more commonly, non-Hodgkin lymphoma can lead to cholestasis [1]. Primary hepatic lymphoma, in the absence of systemic disease, is rare and accounts for <1 % of cases [151]. Forty to 70 % of patients with hepatic disease will have elevations in serum alkaline phosphatase and bilirubin. Diffuse large cell type B-cell lymphoma is responsible for most cases of primary hepatic lymphoma [151]. A more rare form of hepatosplenic T-cell lymphoma may also cause cholestasis in the afflicted patient [1, 152]. Solid organ and HSCT recipients are at risk for post-transplant lymphoproliferative disorder (PTLD) [153, 154]. This Epstein–Barr virus associated type of non-Hodgkin lymphoma can involve the liver and can lead to either intrahepatic or extrahepatic (obstructive) cholestasis [1].
Graft-Versus-Host Disease
GVHD is a major complication following HSCT which can involve the liver, among many other organ systems [2]. Its incidence has been reported in up to 80 % of transplant recipients, depending on the type of transplant [155]. GVHD presents as either acute—within the first 100 days after HSCT—or chronic disease.
Pathogenesis
GVHD develops when donor-derived T-cells expand within the recipient and recognize host antigens as foreign. Several risk factors predispose to the development of GVHD, including HLA mismatch, gender mismatch, reduced-intensity conditioning, and high number of T-cells transported from the donor [156, 157]. Acute GVHD (aGVHD) results directly from the degree of human leukocyte antigen (HLA) mismatch. The donor cytotoxic T-cells target host hepatocytes, epithelial cells of the small bile ducts and periductal glands [158]. Host cell death is mediated by apoptosis via the Fas/Fas ligand pathway. Histologically, there is lymphocytic infiltration and destruction of bile ducts as well as endothelialitis [159, 160]. Apoptotic bodies are seen [159]. Bile duct injury is also typical of chronic GVHD (cGVHD); however, development of fibrosis is rare unless there is concomitant underlying liver disease.
Clinical
Liver involvement in aGVHD generally follows skin involvement, which consists of a maculopapular rash involving the palms and soles. Acute disease is often a diagnosis of exclusion, the differential including sinusoidal obstruction syndrome, viral infection, drug toxicity, and sepsis [155]. The patient develops cholestatic liver injury and subsequent jaundice. Tender hepatomegaly and fluid retention are common and patients may develop fever, nausea, and anorexia [155]. Serum biochemistries will generally show an elevation in conjugated bilirubin, alkaline phosphatase, and gamma-glutamyltranspeptidase, although a more hepatitic presentation with elevations in alanine aminotransferase has been described [155, 161–163]. Coagulopathy and elevated ammonia levels can be seen. Acute GVHD is considered to be severe if the serum bilirubin level is >6.1 mg/dL, and the prognosis is poor in this setting [164]. The grade of aGVHD severity, generally based upon the modified Seattle Glucksberg criteria, carries prognostic significance (Table 11.4) [165–167].
Skin rash | Bilirubin (mg/dL) | Gut fluid loss | |
|---|---|---|---|
Grade I | <25 % of body | 1.52–3.5 | 500–1,000 mL/day |
Grade II | 25–50 % | 3.57–8.0 | 1,000–1,500 mL/day |
Grade III | >50 % or erythroderma | 8.0–15.0 | >1,500 mL/day |
Grade IV | Bullae; desquamation of skin | >15 | >2,500 mL/day or ileus |
Chronic GVHD (cGVHD) develops 100 days or more after transplant. Those who develop aGVHD are more likely to develop cGVHD, but this is not a prerequisite. Additionally, older age is a risk factor for its development [157]. Chronic GHVD can have similar clinical findings to aGVHD or presents with overlap features of both acute and chronic disease [155, 168]. Features which are diagnostic of cGVHD include skin poikiloderma, lichen planus, lichen-type changes on mucus membranes, fasciitis, and joint contractures [155]. Distinctive features include alopecia, nail dystrophy, xerostomia, mucoceles, mouth ulceration, keratoconjunctivitis sicca, and myositis. Liver involvement is evidenced by continued or worsening cholestasis. There may be sudden or progressive elevation in the alkaline phosphatase and gamma glutamyl transpeptidase. The development of hyperbilirubinemia is generally late in the course and coincides with the development of cirrhosis and small bile duct destruction [159]. cGVHD is classified as mild, moderate, or severe (Table 11.5) [168, 169]. The differential diagnosis includes viral infection, and liver biopsy should be performed in this setting to establish the diagnosis. Up to 80 % of patients will show improvement with initiation of immunosuppressive therapy—corticosteroids with or without a calcineurin inhibitor. Additionally, ursodeoxycholic acid may be used. Patients who fail to respond have a poor long-term prognosis [2].
Mild | • Involvement of 1 or 2 organs (not lung involvement) with a score of 1 |
Moderate | • Involvement of 3 organs with a score of 1, or • Involvement of 1 organ with a score of 2, or • Pulmonary involvement with a score of 1 |
Severe | • Major disability in any site, or • Pulmonary involvement with a score of 2 or 3 |
Treatment
The use of prophylactic immunosuppression has been associated with a 20–25 % decreased incidence of aGVHD and subsequently cGHVD [155, 170]. Ursodeoxycholic has also been shown to reduce the incidence of hepatic aGVHD and is generally used prophylactically during the first 80 days following HSCT [171, 172].
Grade I aGVHD rarely requires systemic therapy. Corticosteroids and calcineurin inhibitors (i.e., cyclosporin, tacrolimus) form the backbone of therapy for those with grade II–IV aGVHD [157, 165]. Immunosuppression leads to resolution in about 30–50 % of cases, with long-term survival rates of 60 % in this group [165, 173–177]. Patients who fail first-line therapy may require extracorporeal photopheresis, anti-tumor necrosis factor alpha (anti-TNFα) antibodies, mTOR (mammalian target of rapamycin) inhibitors, mycophenolate mofetil, or interleukin-2 receptor antibodies [165, 178–180]. Third-line agents include mesenchymal stem cells, alemtuzumab (Campath 1H), or pentostatin [165]. Long-term survival in this group drops to only 20–35 % [174]. Patients who fail therapy may go on to develop cGVHD [2]. Therapy is similar to that of aGVHD using systemic corticosteroids. Failing that, second-line therapies include extracorporeal photopheresis, mTOR inhibitors, rituximab, pentostatin, methotrexate, or mycophenolate mofetil.
Sinusoidal Obstruction Syndrome (SOS)
Veno-occlusive disease (VOD) was first described in 1920 following ingestion of Senecio tea containing pyrrolizidine alkaloids [181]. It was first described following HSCT in 1979 [182, 183]. The term veno-occlusive disease has fallen out of favor and has been replaced by sinusoidal obstruction syndrome as a better reflection of its pathophysiology [184, 185]. Development of SOS is related to the conditioning regimen used prior to stem cell infusion (myeloablative HSCT) with a variably reported incidence of 5–70 %, with a mean incidence of about 14 % [2, 186–191]. It has a reported mortality of up to 50 %, averaging 15–20 % [184, 192, 193].
Pathogenesis
The primary inciting event is the randomly distributed development of hepatic sinusoidal endothelial injury usually within zone 3 of the hepatic acinus [184, 186, 194–196]. Loss of endothelial cell fenestrations occurs and gaps in the endothelial cell lining form, ultimately leading to frank endothelial denudation and loss of sinusoidal integrity [197]. Involvement of the hepatic venules is not essential to the diagnosis and is present in 50–75 % of cases [184, 194, 198–200]. There is edematous thickening in the subintimal zone of the central and sublobular venules. This leads to concentric luminal narrowing with subsequent increased resistance to blood flow, resulting in hepatic congestion, sinusoidal dilation, and portal hypertension [184]. The congestion is associated with extravasation of red blood cells into the perisinusoidal space (space of Disse) [198]. Fragmented red blood cells, fibrin, and factor VIII have been shown to be present, and contribute further to partial to complete fibrotic obliteration of the venules [2, 195]. Focal ischemia leads to progressive derangements within the liver, with cytokine release [201, 202]. Hepatocyte atrophy, injury and death occur, prominently localized to the centrilobular region. Injury can be potentiated by medications used in the prophylaxis of GVHD such as sirolimus and tacrolimus [203]. SOS may also be associated with other liver lesions, including peliosis hepatis, nodular regenerative hyperplasia, and centrilobular and venular fibrosis [198, 204–206].
Controversy remains as to whether the coagulation pathways play a role in the pathogenesis of SOS [184, 204]. There is evidence that the damaged endothelium elicits activation of the coagulation cascade [2, 193, 207–210]. Levels of anticoagulant proteins are decreased in patients with SOS, while levels of procoagulant proteins are elevated [209–213]. It remains unclear whether this is cause or effect, as the presence of thrombi in the sinusoids and venules is uncommon [184, 194].
The drugs which lead to SOS show preferential toxicity to sinusoidal endothelial cells rather than other cell types within the liver [214, 215]. The incidence of SOS appears to be higher in patients undergoing allogeneic compared with autologous HSCT, although this may be related more to the type of conditioning regimen [184, 188]. Additional risk factors for the development of SOS include preexisting liver disease, hepatic metastases, advanced age, female gender, unrelated or HLA-mismatched related donors, prior use of vancomycin, acyclovir, norethisterone or gemtuzumab ozogamicin, use of the conditioning agents cyclophosphamide, busulfan, melphalan, and/or high dose total body irradiation over 14 Gy, and concomitant sirolimus use [158, 184, 188, 189, 191, 216–224]. Risk may be cumulative if patients require a second HSCT and were previously exposed to these agents. Cyclophosphamide carries the greatest risk for causing SOS. It is metabolized by cytochrome P450 into its therapeutic metabolite, phosphoramide, and the toxic metabolite, acrolein. Acrolein causes injury to the adjacent endothelial cells which can be ameliorated by glutathione [184, 225–228]. Glutathione is then depleted by over 95 % prior to onset of hepatic toxicity [229]. The use of N-acetylcysteine has shown mixed results. Toxicity is more likely if cyclophosphamide is combined with total body irradiation over 14 Gy [223, 230]. The incidence of SOS can be decreased by fractionating the total body irradiation dosing or increasing the interval between irradiation and cytotoxic therapy [217, 231, 232]. Busulfan given concurrently with cyclophosphamide potentiates its hepatotoxicity [217, 233]. The risk increases when the area under the curve for busulfan is >1,500 μmol/min/L and thus close monitoring of its serum levels is critical [234, 235]. Gemtuzumab ozogamicin is a monoclonal antibody against CD33. It carries a 15–40 % risk of causing sinusoidal injury, particularly if given prior to cyclophosphamide [236]. This antibody was withdrawn from the US market in 2010.
Clinical
Clinical criteria for the diagnosis of SOS have been developed (Table 11.6) [190, 223]. Patients present within the first 3–4 weeks after HSCT with a wide spectrum of severity, ranging from mild, reversible disease to severe often fatal illness (Table 11.7) [2, 184, 192, 193, 199, 200, 223]. Classically, SOS is characterized by rapid weight gain with or without ascites, tender hepatomegaly, and elevated bilirubin/jaundice. Asymptomatic weight gain precedes the other symptoms, occurring within 6–8 days of the transplant in the majority of patients and results from avid renal sodium and water retention. Within days, an isolated elevation in conjugated bilirubin becomes obvious [184]. Elevations in alkaline phosphatase and serum aminotransferases accompany the progressive increase in bilirubin. Patients complain of right upper quadrant pain, which can be severe enough to require narcotic pain relief. Half of those with severe SOS develop hepatorenal syndrome and 25–50 % require hemodialysis [2]. The fluid overload and thrombocytopenia are refractory to therapy [193, 237]. As disease advances, SOS can lead to prolonged prothrombin time, encephalopathy, and/or multiorgan failure.
Table 11.6
Criteria for diagnosis of sinusoidal obstruction syndrome
Baltimore criteria [190] | Modified Seattle criteria [223] |
|---|---|
Within 21 days of transplantation: • Serum bilirubin > 2 mg/dL (34 μmol/L) • At least two of the following: • Hepatomegaly • >5 % weight gain from baseline • Ascites | Within 20 days of transplantation, at least two of the following: • Serum bilirubin >2 mg/dL (34 μmol/L) • Hepatomegaly with right upper quadrant pain • >2 % weight gain from baseline due to fluid retention |
Mild | • No adverse effects of liver disease, and • No medications required for diuresis or hepatic pain, and • All symptoms, signs and laboratory features reversible |
Moderate | • Adverse effects of liver disease present, and • Sodium restriction or diuretics required, or • Medication for hepatic pain required, and • All symptoms, signs and laboratory features reversible |
Severe | • Adverse effects of liver disease present, and • Symptoms, signs or laboratory features not resolved by day +100, or • Death |
Diagnosis
One must rule out other causes of cholestatic liver injury, including aGVHD, cyclosporine-induced hepatotoxicity, fungal infection, viral hepatitis, cholestasis of sepsis, drug-induced cholestasis, PNLD, and persistent tumor infiltration. Hepatic Doppler imaging often shows ascites, hepatomegaly, hepatic vein dilation and, late in the course of disease, evidence of portal hypertension with reversal of venous flow in the portal veins [238]. Magnetic resonance imaging reveals hepatomegaly, hepatic vein narrowing, periportal cuffing, ascites, and signs of reduced portal venous flow velocity [184]. Liver biopsy remains the gold standard for diagnosis, but may not be needed. When performed, it should be via the transjugular route and hepatic venous pressure gradients should be obtained. A gradient of over 10 mmHg has a specificity of >90 % and a positive predictive value of over 85 % for SOS [239, 240]. Elevated serum markers of endothelial injury, such as plasminogen activator inhibitor type-1 (PAI-1), can be measured and may be useful as a diagnostic and prognostic biomarker of SOS [241].
Treatment
Successful management of SOS involves prevention if possible. Alteration of the conditioning regimen and/or the GVHD prophylaxis may decrease the incidence of SOS [241]. The use of prophylactic ursodeoxycholic acid has been shown to reduce the incidence of SOS [171]. Defibrotide has shown some promise in decreasing the incidence of SOS, but large randomized controlled trials are lacking [242]. No clear evidence of preventive benefit has been shown with prostaglandin E1, tissue plasminogen activator, N-acetylcysteine, human antithrombin III concentrate, activated protein C, low-dose heparin, albumin, or prednisone [216, 241, 243, 244]. Low-dose dopamine may be of some benefit in patients with hepatorenal syndrome.
Mild SOS is self-limited, and no treatment is required. Unfortunately, there are no known effective treatments for moderate to severe disease and care remains largely supportive. Severe SOS is heralded by a rapidly rising serum bilirubin and increasing aminotransferase levels, progressing to portal vein thrombosis and multiorgan failure. It carries a mortality rate of over 80 % [186, 193]. The use of systemic anticoagulants and thrombolytics has been mixed, showing no survival advantage, and is associated with significant bleeding complications [216]. Defibrotide has been shown to improve the signs and symptoms of SOS in about 42 % of those afflicted [241, 245].
Summary
Cholestasis in the hospitalized patient has a broad differential given the numerous potential systemic causes [1]. In the HSCT, cholestasis is often an ominous sign of poor outcome, although preventive measures have improved survival. Diagnosis requires a thorough history and evaluation. Patients require aggressive supportive care and removal of the cause of cholestasis if possible.
References
1.
Delemos AS, Friedman LS. Systemic causes of cholestasis. Clin Liver Dis. 2013;17(2): 301–17.PubMed
2.
Tuncer HH, Rana N, Milani C, Darko A, Al-Homsi SA. Gastrointestinal and hepatic complications of hematopoietic stem cell transplantation. World J Gastroenterol. 2012;18(16): 1851–60.PubMedCentralPubMed
3.
Gratwohl A, Baldomero H, Aljurf M, Pasquini MC, Bouzas LF, Yoshimi A, et al. Hematopoietic stem cell transplantation: a global perspective. JAMA. 2010;303(16):1617–24.PubMedCentralPubMed
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


