Several centrally acting agents have shown potential to improve erectile function in men with ED. They still lack adequate data in efficacy and tolerability. Nasal formulations of apomorphine and bremelanotide seem to be the most likely candidates for future approval. They may play a role, specifically in men who fail phosphodiesterase 5 (PDE5) therapy, are unable to take PDE5 inhibitors because of side effects, or are on nitrate therapy. This article reviews the centrally acting agents and the data on their efficacy.
Cortical regions act as centers for integration of sensory stimuli and hormonal influences to initiate sexual desire and libido. These stimuli and hormones then act through sympathetic and parasympathetic pathways to control the peripheral activities that result in a penile erection. These cortical pathways explain the occurrence of erections without genital stimulation such as those occurring during fantasy, visual stimuli, and sleep. Whereas the role of nitric oxide (NO) as an end effector has been well established, the role of the central nervous system (CNS) in mediating penile erections remains unclear despite several laboratory and animal studies attesting to its importance. Initial studies were based on animal models with retrograde labeling of pathways, but more recent reports have used newer techniques such as the positron emission tomographic scan.
Among a large variety of areas that may potentially be involved within the cortex, the medial preoptic area and the paraventricular nucleus (PVN) of the hypothalamus along with the hippocampus seem to be the principal areas of interest. The PVN contains dopaminergic neurons whose stimulation is associated with penile erection. Whereas injection of dopaminergic agents in this region potentiates erections, lesions in this region result in a loss of erectile ability. In further attempts to characterize the role of the PVN in erectile functioning, Richards and colleagues recorded potentials from individual neurons in the PVN as well as local field potential activity in anesthetized rats during erectile activity. Apomorphine in erectogenic doses was injected peripherally and resulted in variable firing patterns of the neurons in the preerectile and erectile phases ( Figs. 1 and 2 ).
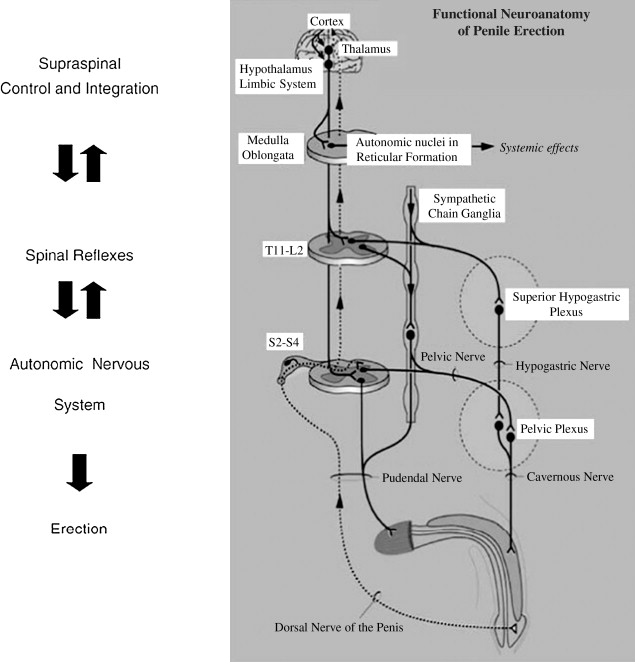
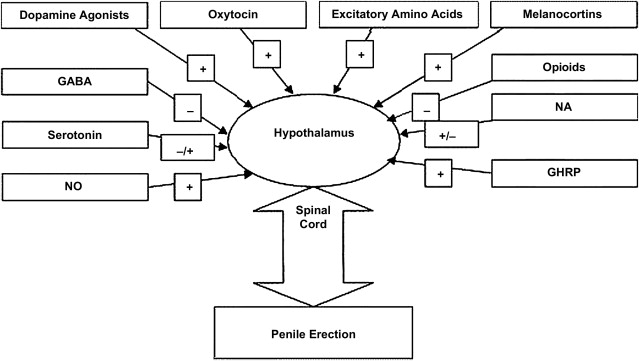
The melanocortinergic system also has multiple sites of action within this complex network. This system consists of neuropeptides such as adrenocorticotropic hormone, β-endorphin, and α, β and γ melanocyte-stimulating hormones (MSHs) apart from their receptors and various antagonists. The melanocortins are posttranslational products of the prohormone pro-opiomelanocortin (POMC), which produces 8 different peptides based on the site of cleavage. POMC messenger RNA exists in several human tissues including the genitourinary tract. Among the various melanocortin receptors (MCRs), MCR3 and MCR4 have been found in the hypothalamus and MCR4 primarily has been seen to be involved in modulating sexual function.
The CNS administration of α-MSH induces penile erections and yawning, somewhat similar to that seen with apomorphine. Mizusawa and colleagues implanted catheters in the lateral cerebral ventricle or the subarachnoid space in 78 male Sprague-Dawley rats and injected α-MSH. These injections resulted in penile erections and an increase in intracavernosal pressure, which was abolished by the administration of NO inhibitors. Similar responses were produced by intracerebroventricular oxytocin, but intrathecal α-MSH did not produce any erectile response, suggesting a central role for α-MSH.
Centrally acting agents are not among the currently recommended treatments for erectile dysfunction (ED) in the guidelines of the American Urological Association and the European Association of Urology. These guidelines recommend phosphodiesterase 5 inhibitors (PDE5i) sildenafil, tadalafil, and vardenafil as first-line therapies with options including prostaglandin E 1 , intracavernosal vasoactive agents, vacuum constriction devices, and penile prosthesis. The guidelines recommended switching from an oral to an alternate therapy among nonresponders.
PDE5i have a significant failure rate, including both primary and secondary failures among patients who may have initially responded to therapy. Further, some patient groups, such as in those after radical prostatectomy, have poorer outcomes with PDE5i. Adverse effects, time to onset of action, and lack of spontaneity are additional concerns with these agents. The current guidelines leave little oral options for these men, and there is clearly a potential for the development of alternative therapies. The principal reason for the recommendation against centrally acting agents is the lack of adequate scientific data on their efficacy and safety. This article reviews the centrally acting agents and the data on their efficacy.
Dopaminergic agents: apomorphine
In one of the earliest clinical reports on the use of apomorphine, Lal and colleagues reported reproducible erections in 7 of 9 men injected subcutaneously with varying doses of the drug. Apart from suggesting the efficacy of apomorphine, this study also showed that these actions were not mediated through cholinergic mechanisms because they were not inhibited by benztropine. Apomorphine has been shown to induce erections even without physical stimulation, solely through visual erotic stimuli. In a randomized blinded study, Danjou and colleagues injected subcutaneous apomorphine in 10 healthy male volunteers at a dose of 9 μg/kg and found that erections were induced starting from the fourth minute after the injection and that this erection was potentiated by visual stimuli in the form of erotic slides.
One of the major hurdles in the use of apomorphine was the lack of an oral formulation. In the pre-PDE5 inhibitor era, this hurdle was a major limitation to the use of drugs in the management of ED. Apomorphine is highly soluble in lipids, has a wide volume of distribution, and undergoes rapid metabolism, thus limiting its actions when administered orally. Although the transmucosal absorption of apomorphine had been documented as early as 1935, the adverse events associated with the large dose limited its clinical usefulness. In 1995, Heaton and colleagues reported a series of experiments using various doses and routes of administration of apomorphine in men without an organic cause of ED. The investigators found excellent outcomes with a sublingual sustained release formulation, which had the least side effects (primarily yawning) when compared with the liquid or oral tablet formulations. After discovery of the sublingual route of administration, several studies evaluated the ideal dose and safety of this preparation. In one of the largest reported studies, 569 men and their partners were enrolled in 1 of 4 arms of a randomized study using dose escalation protocols, fixed doses of apomorphine, or placebo at 51 centers. A total of 444 men completed the 8-week study. A statistically significant higher proportion of men in all the 3 treatment arms attained and maintained an erection compared with the placebo group. The international index of erectile function (IIEF) score in all the treatment groups was higher than the score in the placebo group. Between 30% and 50% patients reported nausea that increased with the dose, but this decreased with increasing period of drug use.
Attempts to formulate an ideal route of administration that minimizes side effects of apomorphine continue. Lu and colleagues showed that intranasal delivery of apomorphine resulted in similar brain and cerebrospinal fluid concentrations of the drug when compared with subcutaneous administration, whereas its plasma concentration was only half. Further, brain concentrations were achieved faster with nasal administration than with subcutaneous injections, leading the investigators to suggest a nose-to-brain pathway for apomorphine. Riley and colleagues recently reported the safety and efficacy of a novel nasal formulation delivered at low doses in men with mild to severe ED. These 2 phase 2b dose-finding studies included a total of 600 patients who were randomized to receive 1 of 3 fixed doses (between 100 and 300 μg) of either apomorphine or placebo on an as-required basis for up to 12 weeks during which they were to attempt intercourse at least 12 times. Changes in the Sexual Encounter Profile (SEP) questions 2 and 3 formed the primary end points. At all tested doses, patients showed an improvement in the ability to achieve vaginal penetration and maintain an erection. The onset of action was within 10 minutes for most responders. The most common adverse effects were headache, nasopharyngitis, and dyspepsia. About 6% of the randomized patients withdrew because of adverse effects. The investigators concluded that nasal apomorphine may become a first-line management for ED because of rapid onset of action and reproducible efficacy.
In direct comparison with existing PDE5i as primary therapy for ED, apomorphine continues to show poorer outcomes. Giammusso and colleagues reported a 20-week, open label, randomized crossover study using flexible dosing. The primary outcome was the sexual domain questions (questions 1–5 and 15) of the IIEF, whereas various efficacy and satisfaction questionnaires formed the secondary end points. Treatment preference was determined at the end of the study among subjects who received both treatments. A total of 130 patients were randomized. In the intent-to-treat analysis, there was significantly greater improvement with sildenafil than with apomorphine. A statistical difference in favor of sildenafil was maintained in the per-protocol analysis. Sildenafil was significantly superior to apomorphine in all secondary parameters evaluated with a much higher events log than apomorphine. It was also superior, significantly, in terms of patient preference. Similar outcomes were reported in a study of 108 patients at 12 centers in Brazil in which sildenafil scored higher in all evaluated parameters. Lack of effectiveness was also reported as the commonest reason for discontinuation of apomorphine in a survey of 11,185 patients in the United Kingdom who had been prescribed this drug as the first-line therapy for ED.
Melanocortins
Several highly potent and selective agonists and antagonists of the melanocortins have been developed to study the effect of this substance on human sexual function. These include melanotan II (MT-II), which is a cyclic peptide analogue of α-MSH and expresses agonistic activity at the MCR4 receptor. PT-141, a metabolite of MT-II, is another synthetic agonist peptide analogue, as is tetrahydroisoquinoline, which has more than 100-fold selectivity for the MCR4 receptor.
The development of erections in men given subcutaneous melatonin for pigmentation disorders led to a single-blind placebo-controlled trial with the superpotent MT-II in 3 normal male volunteers. All subjects noted intermittent penile erections along with stretching and yawning about 1 to 5 hours after the injections. Spurred by these findings, Wessells and colleagues conducted a randomized, controlled, crossover study in 10 men with psychogenic ED. Subcutaneous injections of MT-II were alternated with saline as placebo. A Rigiscan (Timm Medical Technologies, Inc, Eden Prairie, MN, USA) device was used to monitor erections for 6 hours after the injection, with the patient awake during this entire period. Of the 10 men, 8 reported erections within the 6-hour study period after the MT-II injection compared with none after the placebo. The onset of erection varied from 15 to 270 minutes, with 144 minutes being the mean duration of erections.
The investigators continued their evaluation by extending it to men with organic causes of ED. A total of 10 men with an average of 2.2 organic causes for ED were randomized in a double-blind, crossover placebo-controlled study of MT-II (0.025 mg/kg) or placebo. Of the 10 men, 9 reported improved erections on at least 1 of the 2 injections of the drug, with 12 of 19 injections resulting in an erection compared with 1 of 21 of the placebo. The erections occurred for an average of 64 minutes. The Rigiscan device confirmed the subjective findings. Further, the investigators noted an increased desire after 10 of the drug injections. Nausea was the commonest side effect in the drug group.
Based on these initial reports, PT-141 (bremelanotide), a synthetic peptide analogue of α-MSH was developed as an agonist to melanocortin receptors, including MCR4. Its systemic administration in rats activates neurons in the hypothalamus and in normal men results in a dose-dependent increase in erectile activity.
Rosen and colleagues reported the results of 2 simultaneous studies using subcutaneous PT-141 in healthy men and sildenafil nonresponders. In the first study, a phase 1 evaluation, 48 healthy men received varying doses of PT-141 or placebo, whereas in the second phase 2a study, fixed doses of PT-141 or placebo were administered in a crossover, blinded, randomized manner to men with ED. Healthy men were not provided visual stimulation, whereas men with ED were. Rigiscan-monitored erectile responses were statistically significant in both groups of PT-141–treated men.
Subsequently, an intranasal formulation of PT-141 was developed and tested in healthy male subjects and in patients with sildenafil-responsive ED. Using a Rigiscan, with or without sexual stimulation, Diamond and colleagues reported a statistically significant erectile response in men receiving PT-141 compared with those receiving placebo.
Salvage of PDE5 inhibitor failures is one of the most likely scenarios for the use of melanocortin agents. Safarinejad and Hosseini reported a randomized controlled trial using bremelanotide, 10 mg, as an intranasal spray, 45 minutes to 2 hours before sexual intercourse, versus placebo in 342 men who failed to respond to sildenafil even after a reeducation program. The patients were asked to assess at least 16 attempts with the therapy. Most men had an organic cause for their ED. About 33% men who took bremelanotide responded to treatment compared with 8% taking placebo. Around 86% patients achieved erections within an hour of treatment, with the mean duration of rigidity sufficient for penetration being more than 10 minutes in men with even severe ED. The mean frequency of intercourse increased to 2.2 per week from 1.2 at baseline in the bremelanotide group. Around 16.3% patients on bremelanotide reported adverse effects with nausea, flushing, and sweating being the commonest.
Melanocortins
Several highly potent and selective agonists and antagonists of the melanocortins have been developed to study the effect of this substance on human sexual function. These include melanotan II (MT-II), which is a cyclic peptide analogue of α-MSH and expresses agonistic activity at the MCR4 receptor. PT-141, a metabolite of MT-II, is another synthetic agonist peptide analogue, as is tetrahydroisoquinoline, which has more than 100-fold selectivity for the MCR4 receptor.
The development of erections in men given subcutaneous melatonin for pigmentation disorders led to a single-blind placebo-controlled trial with the superpotent MT-II in 3 normal male volunteers. All subjects noted intermittent penile erections along with stretching and yawning about 1 to 5 hours after the injections. Spurred by these findings, Wessells and colleagues conducted a randomized, controlled, crossover study in 10 men with psychogenic ED. Subcutaneous injections of MT-II were alternated with saline as placebo. A Rigiscan (Timm Medical Technologies, Inc, Eden Prairie, MN, USA) device was used to monitor erections for 6 hours after the injection, with the patient awake during this entire period. Of the 10 men, 8 reported erections within the 6-hour study period after the MT-II injection compared with none after the placebo. The onset of erection varied from 15 to 270 minutes, with 144 minutes being the mean duration of erections.
The investigators continued their evaluation by extending it to men with organic causes of ED. A total of 10 men with an average of 2.2 organic causes for ED were randomized in a double-blind, crossover placebo-controlled study of MT-II (0.025 mg/kg) or placebo. Of the 10 men, 9 reported improved erections on at least 1 of the 2 injections of the drug, with 12 of 19 injections resulting in an erection compared with 1 of 21 of the placebo. The erections occurred for an average of 64 minutes. The Rigiscan device confirmed the subjective findings. Further, the investigators noted an increased desire after 10 of the drug injections. Nausea was the commonest side effect in the drug group.
Based on these initial reports, PT-141 (bremelanotide), a synthetic peptide analogue of α-MSH was developed as an agonist to melanocortin receptors, including MCR4. Its systemic administration in rats activates neurons in the hypothalamus and in normal men results in a dose-dependent increase in erectile activity.
Rosen and colleagues reported the results of 2 simultaneous studies using subcutaneous PT-141 in healthy men and sildenafil nonresponders. In the first study, a phase 1 evaluation, 48 healthy men received varying doses of PT-141 or placebo, whereas in the second phase 2a study, fixed doses of PT-141 or placebo were administered in a crossover, blinded, randomized manner to men with ED. Healthy men were not provided visual stimulation, whereas men with ED were. Rigiscan-monitored erectile responses were statistically significant in both groups of PT-141–treated men.
Subsequently, an intranasal formulation of PT-141 was developed and tested in healthy male subjects and in patients with sildenafil-responsive ED. Using a Rigiscan, with or without sexual stimulation, Diamond and colleagues reported a statistically significant erectile response in men receiving PT-141 compared with those receiving placebo.
Salvage of PDE5 inhibitor failures is one of the most likely scenarios for the use of melanocortin agents. Safarinejad and Hosseini reported a randomized controlled trial using bremelanotide, 10 mg, as an intranasal spray, 45 minutes to 2 hours before sexual intercourse, versus placebo in 342 men who failed to respond to sildenafil even after a reeducation program. The patients were asked to assess at least 16 attempts with the therapy. Most men had an organic cause for their ED. About 33% men who took bremelanotide responded to treatment compared with 8% taking placebo. Around 86% patients achieved erections within an hour of treatment, with the mean duration of rigidity sufficient for penetration being more than 10 minutes in men with even severe ED. The mean frequency of intercourse increased to 2.2 per week from 1.2 at baseline in the bremelanotide group. Around 16.3% patients on bremelanotide reported adverse effects with nausea, flushing, and sweating being the commonest.
Related posts:
 Penile Rehabilitation After Prostate Cancer Treatment: Outcomes and Practical Algorithm
Testosterone and Prostate Cancer: What are the Risks for Middle-Aged Men?
Psychological Factors Associated with Male Sexual Dysfunction: Screening and Treatment for the Urologist
Priapism: New Concepts in Medical and Surgical Management
Penile Rehabilitation After Prostate Cancer Treatment: Outcomes and Practical Algorithm
Testosterone and Prostate Cancer: What are the Risks for Middle-Aged Men?
Psychological Factors Associated with Male Sexual Dysfunction: Screening and Treatment for the Urologist
Priapism: New Concepts in Medical and Surgical Management
 Testosterone Deficiency and Risk Factors in the Metabolic Syndrome: Implications for Erectile Dysfunction
Peyronie’s Disease: Review of Nonsurgical Treatment Options
Testosterone Deficiency and Risk Factors in the Metabolic Syndrome: Implications for Erectile Dysfunction
Peyronie’s Disease: Review of Nonsurgical Treatment Options
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


