The human microbiome is a new frontier in biology and one that is helping to define what it is to be human. Recently, we have begun to understand that the “communication” between the host and its microbiome is via a metabolic superhighway. By interrogating and understanding the molecules involved we may start to know who the main players are, and how we can modulate them and the mechanisms of health and disease.
Key points
- •
The human microbiome is a new frontier in biology and one that is helping to define what it is to be human.
- •
Recently, we have begun to understand that the “communication” between the host and its microbiome is via a metabolic superhighway.
- •
By interrogating and understanding the molecules involved, we may start to know who the main players are, and how to modulate them and the mechanisms of health and disease.
Introduction
Understanding mammalian biology has for the best part of 100 years been focused on trying to model how this system interacts with the environment. Since the discovery that DNA contains all the necessary information to recreate a new living organism and coupled with the revolution in gene sequencing, this focus turned to try to understand how host’s genome interacts with its environment to influence the balance between health and disease. A significant component of this body of work has been focused on the role of microbial pathogens in driving disease phenotypes. However, there is a dearth of information that considers that the microbes colonizing the various niches of the human body may actually have coevolved with the host and provide essential functions not found in the host’s genome. The role of mammalian microbiome is revealed, in extremis, when animals are reared in a sterile environment and thus do not develop in the presence of a microbiota. The absence of the microbiome has been shown to influence a very wide and disparate range of physiologic parameters, including cardiac size and output, response to anesthesia, and many other features of the mature mammalian system. Although we can see a fundamental role for the microbiome in the development of a mature host, we are left with a dearth of mechanisms by which this process is driven.
Introduction
Understanding mammalian biology has for the best part of 100 years been focused on trying to model how this system interacts with the environment. Since the discovery that DNA contains all the necessary information to recreate a new living organism and coupled with the revolution in gene sequencing, this focus turned to try to understand how host’s genome interacts with its environment to influence the balance between health and disease. A significant component of this body of work has been focused on the role of microbial pathogens in driving disease phenotypes. However, there is a dearth of information that considers that the microbes colonizing the various niches of the human body may actually have coevolved with the host and provide essential functions not found in the host’s genome. The role of mammalian microbiome is revealed, in extremis, when animals are reared in a sterile environment and thus do not develop in the presence of a microbiota. The absence of the microbiome has been shown to influence a very wide and disparate range of physiologic parameters, including cardiac size and output, response to anesthesia, and many other features of the mature mammalian system. Although we can see a fundamental role for the microbiome in the development of a mature host, we are left with a dearth of mechanisms by which this process is driven.
Why do we need to know what metabolites are made?
The history of microbiology has been focused predominantly on understanding the role that pathogens play in disease, and this goes back to the time of Robert Koch and Louis Pasteur. However, in the last 15 years there has been a slow but inexorable move toward understanding how the commensal and mutualistic members of the human microbiome also contribute to host health and disease initiation. In the last 5 years, this interest in the microbiome has really expanded at an exponential rate. However, we cannot treat these organisms in a similar fashion to pathogenic microbes, because they have not evolved specific strategies to invade, colonize and reproduce in a hostile environment. Many of the functions and features that they possess, and on which we rely, do not conform to the virulence model that we have used to describe and understand pathogens. Many of the functions are actually part of that everyday metabolism of these organisms and as such cannot be considered as virulence factors. For example, for many anaerobic bacteria that colonize the large intestine the ability to ferment simple molecules, to extract energy from them, results in a wide range of metabolites that are bioactive and interact with a wide range of receptors within the host. Thus, the communication between this diverse set of organisms and its host is predominantly via a metabolite superhighway. Thus, to understand this communication we need to be able to characterize the wide array of metabolites that the bacteria produce in response to the environment in which they find themselves and understand how the host responds to these metabolites based on the genes that they have.
How do assess them and what can we assess: Nuclear MR and mass spectrometry, what samples?
Microbial metabolites are typically present in feces, luminal contents and blood, particularly the hepatic portal vein blood, whereas host–microbial cometabolites are present more commonly in circulating blood and urine. Metabolic profiling approaches are increasingly used to study metabolic function of the gut microbiota. The practical implementation of metabolic profiling includes 5 steps: (1) sample collection and preparation, (2) biochemical composition analyses, (3) data analysis and integration (eg, statistically correlating metabolic and microbial data), (4) biomarker recovery and identification, and (5) validation and application.
Urine and blood plasma or serum collection is straightforward, whereas obtaining fecal samples is more challenging and rarely done at outpatient clinics. Moreover, fecal samples are complex in nature because they contain microbial and mammalian cells and food residues, in which the biological and chemical processes continue during postvoiding and sample handling. Hence, storing samples at a lower temperature and immediate sample processing reduce the variation induced by sample handling. Standard operating procedures for biofluid collection and the effects of various handling conditions on the biochemical composition have been reported previously. Analytical platforms, including nuclear MR (NMR) spectroscopy and mass spectrometry (MS), are commonly used in metabolic profiling and can detect a wide range of microbial metabolites and host–microbial cometabolites. NMR spectroscopy is a robust analytical platform with high reproducibility and it generates the most easily accessible and comprehensive information on metabolite structures. Although the sensitivity of NMR spectroscopy is less than MS, it is nondestructive and requires minimum sample preparation. A single proton ( 1 H) NMR experiment using a 600 MHz NMR spectrometer takes about 5 to 10 minutes and can detect a wide range of metabolites including amino acids, fatty acids, phenols, indole, and other organic acids containing protons at low micromolar levels. Therefore, it serves as the first choice for global profiling. MS provide complementary molecular information (eg, molecular mass) and it is much more sensitive than NMR spectroscopy, but often requires preseparation techniques such as liquid chromatography (LC) and gas chromatography (GC). Depending on the metabolites of the interest, different methods can be used in LC to focus on subsets of molecules. For example, reversed phase chromatography is used to study nonpolar compounds, whereas hydrophilic interaction LC is used for detecting polar compounds. Both reversed phase LC-MS and hydrophilic interaction LC-MS are routinely used to analyze the same sample sets to achieve wider metabolite coverage. GC-MS is also a sensitive tool in metabolic profiling and commonly used to quantify short chain fatty acids. However, the drawback of GC-MS is that it requires derivatization of the samples, a long sample preparation procedure, and only volatile compounds or compounds that are volatile after derivatization can be detected. The main metabolic profiling platforms and their strengths and limitations have been summarized by Holmes and colleagues in 2015. Detailed experimental protocols for global metabolic profiling and bile acid profiling have been published as well.
All of these analytical tools generate signal-rich data, which requires multivariate statistical analyses to extract useful information from the datasets. Multivariate data analysis methods, typically including principal component analysis, orthogonal projections to latent structures-discriminant analysis, and random forest, provide easy visualization of the metabolic similarities and differences between the samples or spectral data. Orthogonal projections to latent structures regression analysis is also used to statistically correlate metabolic data with other types of datasets, such as body weight, histologic scores, bacterial counts generated from 16S ribosomal RNA gene-based sequencing platform, cytokines, toxicity ( Fig 1 for an example). Such correlation analysis between metabolic and microbial datasets allows further insight on metabolites that are likely to be associated with gut microbial composition. The statistical modeling results in a panel of spectral signals that are important for class discrimination (eg, treatment group vs control patients). Signal or feature identification can be challenging in global metabolic profiling. There are many publicly available databases such as human metabolome database and METLIN, software including Chenomx NMR Suite (Chenomx Inc, Edmonton, Ontario, Canada) and AMIX (Bruker, Billerica, MA) and published literature sources, that can assist in providing metabolite candidates for the selected features. Statistical total correlation spectroscopy analysis is a statistical tool to calculate correlation between the peaks from the same molecules or the same biological pathways. Further analytical experiments should be carried out to confirm the metabolite identification. Various 2-dimensional NMR spectroscopic experiments can be used to elucidate the connectivity of protons and carbons of the metabolites. Tandem MS/MS can be used to obtain fragmentation patterns of the selected MS features to provide submolecular information for metabolite identification. In the case of targeted signals or metabolites at very low concentrations, solid phase extraction is often used to separate the signals of interest and concentrate it up for further 2-dimensional NMR experiments. In addition, metabolite candidates can be confirmed by spiking the standard compounds in the original biological samples and being tested by NMR spectroscopy or comparing the LC retention times and MS fragmentation patterns from the standards and the samples. Metabolite identification is a time-consuming step and is considered to be a bottle neck in the metabolic profiling approach. These metabolite identification methods are often combined to elucidate the structure of the targeted spectral signals. Approximate numbers of metabolites seen in different biofluids can be in the range of thousands for both urine and serum. Statistical validation can be carried out using methods such as n -fold cross-validation and permutation testing, whereas biological validation remains challenging owing to further requirement of knowledge of the target metabolic pathways, appropriate validation approaches and additional resources. Statistically and biologically validated output from metabolic profiling may eventually be applied to further mechanistic investigation, and clinical diagnosis and therapeutic decision making.
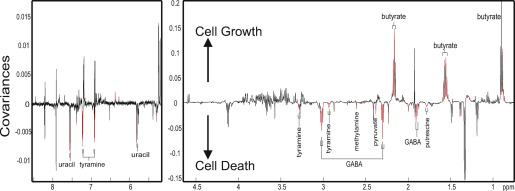
Examples of using metabolic profiling to study gut microbial functionality
The advancement of systems biology techniques, in particular metabolic profiling (metabolomics/metabonomics) and mathematical modeling approaches, has expanded the resolution at which we can study the metabolic contribution of the gut microbiota and their interaction with host biochemistry. A key strength of metabolic profiling is its holistic nature, simultaneously capturing vast amounts of metabolic information without bias, surpassing the need for a specific hypothesis allowing open questions to be asked. This property is ideal for studying the gut microbiota owing to its megavariate host-specific nature and our relatively limited understanding. Instead, metabolic profiling is a hypothesis-generating top-down approach that can illuminate linkages between the gut microbiota and host metabolic pathways for further evaluation.
Coupling these data-rich techniques with gnotobiotic (or germ-free or sterile) and antibiotic-treated animal models has allowed these biochemical associations to be elucidated and their relevance to health and disease to be studied. Pair-wise comparisons of the plasma metabolic phenotypes between gnotobiotic and conventionalized mice using an LC-MS and GC-MS–based approach highlighted the influential role of the gut microbiota on circulating amino acids and organic acids. Differences were observed in the plasma levels of bioactive indole-containing metabolites derived from tryptophan such as indoxyl sulfate and indole-3-propionic acid. The absence of these metabolites in the gnotobiotic animals, coupled with their greater abundance of tryptophan, indicates that this tryptophan metabolism depends on the gut microbiota. Certain bacteria possess tryptophanase activity (a deamination of the amino acid) and can break down dietary tryptophan to indole. This molecule can be absorbed from the gut and metabolized in the liver to indoxyl before being sulphated to indoxyl-sulphate. Indole can also be further processed by a different set of intestinal bacteria to the antioxidant indole-3-propionic acid. The plasma of gnotobiotic animals also contained greater amounts of the amino acid tyrosine, whereas the conventional plasma contained greater amounts of the microbial-host cometabolite 4-cresyl-sulphate. Intestinal bacteria have been shown to metabolize dietary tyrosine to 4-cresol that, upon absorption from the gut, is sulphated in the liver to 4-cresyl sulfate ( p -cresyl sulfate). These findings demonstrate the influence of the gut microbiota on the bioavailability of dietary amino acids, precursors for a range of essential bioactive metabolites.
Similarly, an 1 H NMR spectroscopy-based metabonomic approach was used to characterize the changes in the urinary metabolic profiles of gnotobiotic rats during 21 days of microbial colonization. Here, the acquisition of the gut microbiota was accompanied by marked changes in the urinary biochemical profile. Elevations were noted in the excretion of hippurate, phenylacetylglycine, and 3- and 4-hydroxyphenylpropionic acid. These are microbial–host cometabolites that result from the microbial metabolism of dietary components. Phenylacetylglycine arises from the bacterial metabolism of the amino acid phenylalanine to phenylacetate, which is conjugated with glycine in the rat liver to form phenylacetylglycine and with glutamine in the human liver to form phenylacetylglutamine. Hippurate is the glycine conjugate of benzoic acid that can be derived from the bacterial metabolism of phenylalanine, chlorogenic acid, and catechins. These molecules can be obtained from a range of polyphenolic compounds found in dietary components such as fruit, vegetables, tea, and coffee. Interestingly, in a large-scale metabolic phenotyping study in humans from China, Japan, the United Kingdom, and the United States, hippurate excretion was found to be inversely associated with blood pressure, a major risk factor for cardiovascular disease. Formate, a product of gut microbial fiber fermentation, was also inversely associated with blood pressure. Another metabonomic study characterized the systemic metabolic adaptation to gut colonization in gnotobiotic mice. After 5 days of conventionalization, the metabolic strategy of the liver shifted from glycogenesis to lipogenesis. This observation was consistent with another study combining a transcriptomic and metabonomic approach to study metabolic response to colonization in the mouse jejunum. Here, 2 days of colonization resulted in the suppression of lipid catabolism (eg, β-oxidation) in the jejunum and activation of anabolic pathways (eg, lipogenesis, nucleotide synthesis, and amino acid synthesis). Such biochemical reorientations occurred in parallel with a rapid increase in body weight. These observations indicate the intimate biochemical relationship between the gut microbiota and host and how the host metabolic phenotype is shaped with the development of the gut microbiota.
Antibiotic-treated animal models offer another tool for investigating microbial–host interactions. Gnotobiotic animals differ phenotypically from conventional animals raised in the presence of bacteria. Gnotobiotic animals have a reduced body weight, a lower metabolic rate, underdeveloped gut structure and absorptive capacity, and an immature immune system, and as such can obscure the interpretation of results. Administering antibiotics to conventionally raised animals allows the influence of the gut microbiota on host biochemistry to be studied while preserving the conventional phenotype. This influence was demonstrated by administering the broad-spectrum antibiotics streptomycin and penicillin in the drinking water of rats for 8 days and in an early study vancomycin to mice. Swann and colleague used 1 H NMR spectroscopy to compare the urinary and fecal metabolic profiles of control, antibiotic suppressed and a group undergoing recolonization (4 days of antibiotics followed by 4 days of control treatment). In this study, antibiotic-induced suppression of the intestinal microbiota reduced the urinary excretion of hippurate, phenylpropionic acid, phenylacetylglycine, indoxyl-sulphate, trimethylamine- N -oxide (TMAO), and the short chain fatty acid, acetate. The excretion of the amino acids taurine and glycine, and the tricarboxylic acid cycle intermediates, citrate, 2-oxoglutarate, and fumarate was increased after microbial attenuation. In addition, all the short chain fatty acids (acetate, butyrate, propionate) were reduced in the feces of the antibiotic-treated rats. Short chain fatty acids arise from the bacterial fermentation of carbohydrates, including nondigestible polysaccharides. Because these products provide a significant energy source for the host, this represents a key function of the gut microbiota salvaging energy from the diet. A human study by Claesson and colleagues correlated fecal metabolic and microbial profiles to highlight a putative statistical association between butyrate and the presence of Ruminococcus or Butyricicoccus . Microbial and metabolic profiling of the recolonizing animals revealed a cage-dependent bacterial recolonization. This difference was mirrored by cage-dependent differences in the metabolic signatures. This highlights the potential for environmental pressures to shape the gut bacterial reestablishment after antibiotic therapy, with downstream implications on the metabolic state of the host.
In addition to global profiling of low-molecular-weight metabolites, we can also target specific molecules or families of molecules, for example, bile acids and eicosanoids. Targeted profiling of the bile acid signature enables a detailed overview of the enterohepatic circulation to be gained and the influence of the gut microbiota to be studied. The circulating and hepatic bile acid pool contains more than 30 known bile acids and the gut microbiota is responsible for driving the majority of this diversity. Primary bile acids (cholic acid and chenodeoxycholic acid) are synthesized in the liver from cholesterol and are conjugated with either taurine or glycine before secretion into the bile. Upon ingestion of a meal, bile acids stored in the gallbladder are expelled from the gallbladder into the small intestine and although the majority are actively absorbed in the small intestine a minor amount (1%–5%; 200–800 mg/d in humans) reaches the colon. It is here that bile acids are modified by the resident microbiota. Many bacteria possess bile salt hydrolase enzymes that deconjugate the bile acid from its amino acid. Once deconjugated, further bacterial modifications can occur such as dehydroxylation giving rise to secondary bile acids such as deoxycholic acid and lithocholic acid. Modified bile acids can be absorbed and recycled to the liver, where they are reconjugated and secreted into the bile. This absorption forms the enterohepatic circulation whereby molecules are shuttled between the host liver and the microbiome. Although bile acids have a key role in lipid digestion and absorption, they are now also recognized as important signaling molecules serving as ligands for the nuclear receptor; farnesoid X receptor, and the plasma membrane–bound G-protein–coupled receptor, TGR5. Through binding to these receptors bile acids can regulate genes involved in lipid and glucose metabolism and energy homeostasis. Using a parallel transcriptomic and metabonomic approach the influence of the gut microbiota on the enterohepatic circulation and its signaling capacity was studied. An LC-MS–based approach identified pronounced variation in the bile acid signatures of conventional and gnotobiotic rats with similar modulations induced by antibiotic treatment. The absence or attenuation of the gut microbiota shifted the bile acid signature to one dominated by taurine-conjugated bile acids and strikingly reduced the diversity of the bile acid pool. Such modulations impacted on the signaling function of the bile acid profile with significant alterations in the expression of genes and pathways regulated by bile acids. In addition to being measured in the blood and liver, bile acids were also measured in tissues outside of the enterohepatic circulation (kidney, heart), indicating a broader signaling role of these microbial–host cometabolites.
Metabolic profiling strategies applied to human studies have also expanded our understanding of the gut microbial contribution to host digestion and metabolism. This is well illustrated by the microbial metabolism of dietary choline to trimethylamine (TMA). Choline is predominantly derived from phosphatidylcholine found in animal sources in the diet. The microbial metabolism of choline involves the cleavage of the C–N bond to liberate TMA and acetaldehyde. Whereas acetaldehyde undergoes further microbial metabolism to ethanol, TMA is absorbed from the gut and oxidized in the liver to form TMAO by the flavin-containing monooxygenase 3 enzyme. TMA can also be demethylated to dimethylamine both endogenously and by the gut microbiota (PMID: 4091797). Microbial processing of choline is well established and TMA and TMAO have been previously observed in biofluids from gnotobiotic and antibiotic-treated rodents. However, recent work in humans has linked this activity to increased cardiovascular disease risk. In a global metabolic profiling study in humans, Wang and colleagues found that 3 plasma metabolites were predictive of increased cardiovascular disease—namely, choline, its metabolite betaine, and TMAO. The role of these metabolites in increased cardiovascular disease risk was investigated by feeding them individually to mice. Both choline and TMAO were found to promote atherosclerosis and all 3 metabolites upregulated the expression of macrophage scavenger receptors known to contribute to the atherosclerotic process. The essential role of the gut microbiota in potentiating the bioactivity of choline through TMA production was confirmed using gnotobiotic mice. In a metabolic profiling study, microbial choline metabolism has also been shown to exacerbate nonalcoholic fatty liver disease, a condition caused by choline deficiency in mice.
The potential for the gut microbiota to influence host drug metabolism has been demonstrated in a human study characterizing the metabolic fate of paracetamol/acetaminophen. The metabolic output of the gut microbiota, specifically 4-cresol, was found to influence the phase II detoxification of this widely used analgesic; 4-cresol has toxic properties and requires detoxification by the host. The primary route of this detoxification is sulfation (both in the gastrointestinal tract and in the liver) before excretion in the urine. This is also the preferred route of detoxification for acetaminophen and both molecules are sulphated by the same human cytosolic sulfotransferase, SULT1A1. Because these 2 molecules compete for binding sites as well as for sulfate, the 4-cresol output of the gut microbiota can influence the ability of the host to sulfate acetaminophen. Alternative routes of detoxification including glucoronidation and phase I metabolism by the cytochrome P450 enzymes. Importantly, phase I metabolism results in the generation of the toxic intermediate, N -acetyl- p -benzoquinone imine. In this study, individuals excreting high amounts of 4-cresol before receiving a standard dose of acetaminophen were found to excrete lesser amounts of acetaminophen sulfate and greater amounts of acetaminophen glucuronide. Such an observation is not limited to acetaminophen and many xenobiotics are detoxified via sulfation. Interestingly, using a molecular epidemiology approach we have also observed 4-cresyl sulfate excretion to be positively correlated with age. This observation was found in both US and Taiwanese populations, suggesting that this age-associated change in the metabolic functionality of the gut microbiome is independent of diet and cultural influences. These data have particular relevance given the greater use of drug therapy with aging.
The influence of the gut microbiota on idiosyncratic drug responses has also been demonstrated in a rodent study with the hepatotoxin, hydrazine. In this metabolic profiling study, the protective effect of an established microbiome was demonstrated in rats with gnotobiotic animals showing a marked toxic response to a typically subtoxic dose. These studies demonstrate the potential of using a global metabolic profiling approach to characterize the metabolic functionality of the gut microbiota to predict the efficacy and safety of orally administered xenobiotics. This represents a step toward a precision medicine approach tailoring pharmacologic interventions to the metabolic status of the complete biological system including contributions from the host genome and the microbiome.
Related posts:
 From Culture to High-Throughput Sequencing and Beyond
Fecal Microbiota Transplantation
Gut Microbiota and Complications of Liver Disease
Impact of Antibiotics on Necrotizing Enterocolitis and Antibiotic-Associated Diarrhea
From Culture to High-Throughput Sequencing and Beyond
Fecal Microbiota Transplantation
Gut Microbiota and Complications of Liver Disease
Impact of Antibiotics on Necrotizing Enterocolitis and Antibiotic-Associated Diarrhea
 The Gut Microbiota in Inflammatory Bowel Disease
Gut Microbiota and Complications of Liver Disease
The Gut Microbiota in Inflammatory Bowel Disease
Gut Microbiota and Complications of Liver Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


