Biologic Therapies in Inflammatory Bowel Disease
Anis Ahmadi
John F. Valentine
INTRODUCTION
The advent of biologic therapy has greatly accelerated the understanding and therapy of inflammatory bowel disease (IBD) and other diseases. The first biologic agent approved by the U.S. Food and Drug Administration (FDA) for use in IBD was infliximab in 1998. Since that time there has been substantial advancement in biologic therapies for the management of IBD, and numerous biologic agents are currently in trials.
Biologic therapies for IBD target specific pathways involved in the immune response and down regulate or prevent T-cell activation, induce T-cell apoptosis, interfere with leukocyte trafficking, and have other effects on the immune system that are still being unraveled. This chapter reviews current available biologic agents and those in advanced stages of clinical evaluation. As biologic agents become available and new data are obtained, the “when and how” to use biologic therapy has to be continuously re-evaluated and requires conscientious consideration of effectiveness, safety and cost, as well as the implications for future treatment options, bearing in mind that Crohn’s disease (CD) and ulcerative colitis (UC) are lifelong diseases. Current recommendations are to avoid use of combined therapy with two biologics. More controversial is the need, or lack thereof, for combined immunomodulator therapy along with a biologic agent. Data to help guide therapy on this issue will be forthcoming and will require careful appraisal.
CLASSIFICATION OF BIOLOGIC THERAPIES
Biologic therapies are divided into five classes: (i) natural or modified preparations of biologic origin such as blood products and vaccines consisting of live, attenuated, or dead organisms; (ii) recombinant peptides or proteins, such as erythropoietin, growth hormone, or cytokines; (iii) antibody-based therapies; (iv) nucleic acid-based therapies; and (v) cellular and gene therapies (1). A complete nomenclature has been established for monoclonal antibodies that end in the suffix “mab,” which gives some indication as to the origin and proposed mechanisms of the antibodies as well as other traits (2). Briefly, generic names containing the prefix “xi” in front of “mab,” such as infliximab or rituximab, denote chimeric products, whereas those containing “zu,” such as natalizumab, denote humanized products, and those without a “z” but still containing the “u,” such as adalimumab, are fully human.
TUMOR NECROSIS FACTOR-ALPHA ANTAGONISTS
Tumor necrosis factor-alpha (TNF-α) is a 51-kd trimeric cytokine consisting of three 17-kd inactive monomers (3). It exerts its proinflammatory property via a series of intracellular events that ultimately activate two major transcription factors, nuclear factor-κB and c-Jun. These activated transcription factors, in turn, trigger genes responsible for the components of the inflammatory response (4). The importance of TNF-α in the pathogenesis of IBD was not appreciated until dramatic responses were observed in CD patients treated with infliximab. We will discuss
three TNF-α antagonists (infliximab, adalimumab, and certolizumab pegol) and review their safety profile. The current data suggest that there is little difference in the efficacy (remission or response) between these anti-TNF agents in CD. The response and remission rates for infliximab in ACCENT I, adalimumab in CHARM, and certolizumab pegol in PRECiSE 2 trials are shown in Figure 8.1.
three TNF-α antagonists (infliximab, adalimumab, and certolizumab pegol) and review their safety profile. The current data suggest that there is little difference in the efficacy (remission or response) between these anti-TNF agents in CD. The response and remission rates for infliximab in ACCENT I, adalimumab in CHARM, and certolizumab pegol in PRECiSE 2 trials are shown in Figure 8.1.
Infliximab
Infliximab (Remicade; Centocor, Malvern, Pennsylvania) is a chimeric (75% human, 25% murine) monoclonal immunoglobulin G1 (IgG1) antibody against human TNF-α that binds to both soluble and membrane-bound TNF-α (5). Binding of infliximab to soluble TNF-α removes a potent proinflammatory signal to macrophages and T cells. Binding of infliximab to membrane-bound TNF, on the other hand, can induce T-cell apoptosis as well as antibody-dependent and complement-dependent cytotoxicity (6). The median half-life of infliximab is 7.7 to 9.5 days at the 5 mg/kg dose (7).
The efficacy of infliximab for induction of clinical response and remission in CD has been well documented. The ACCENT I trial in nonfistulizing CD published in 2002 demonstrated that 58% of CD patients responded to a single infusion of infliximab, with 39% and 45% (5 and 10 mg/kg doses, respectively) remaining in remission at week 30, and similar results at week 54 when infliximab therapy was maintained every 8 weeks (8). The ACCENT II trial demonstrated that infliximab at 5 mg/kg every 8 weeks (after an induction regimen) was also efficacious at treating fistulas, with complete fistula closure in 36% of patients treated for 54 weeks (9).
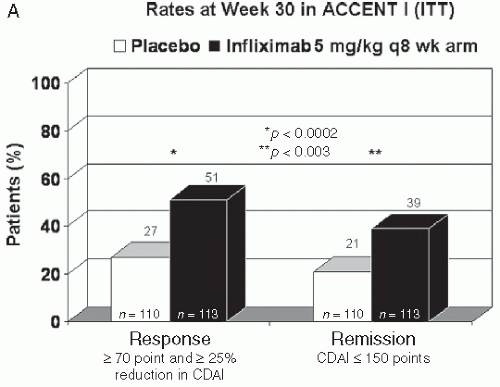 FIGURE 8.1 Response and remission rates in ACCENT I (A), CHARM (B), and PRECiSE 2 (C) trials (8,14,18) are shown. In all three trials, patients received open-label induction, and responders at 2 weeks for infliximab and at 4 weeks for adalimumab and certolizumab pegol were randomized to either placebo or ongoing treatment with the doses shown. These trials were similarly designed; however, it is important to realize that head-to-head comparison studies do not exist and that the patient groups although similar were not the same. |
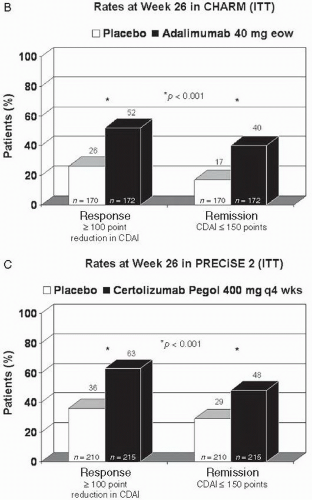 FIGURE 8.1 (Continued) |
Although initial pilot studies suggested that infliximab had minimal therapeutic effect in UC, the ACT I and ACT II trials proved otherwise. These large (364 UC patients in each trial), randomized controlled trials demonstrated that an induction regimen of infliximab 5 mg/kg (at weeks 0, 2, and 6) was effective in inducing a clinical response (69%—ACTI, and 64%—ACT2) and clinical remission (39%—ACT 1,
and 34%—ACT 2) in patients with moderate-to-severe UC at week 8. In addition, a significantly greater proportion of patients receiving infliximab demonstrated mucosal healing and were able to discontinue corticosteroids compared to patients receiving placebo (10). The efficacy of infliximab in hospitalized patients that have failed intravenous (IV) corticosteroids has thus far not been impressive and appears less effective than cyclosporine in this setting. However, there may be a role for those that are unable to take azathioprine or 6-mercaptopurine, a group in which cyclosporine would usually not be considered.
and 34%—ACT 2) in patients with moderate-to-severe UC at week 8. In addition, a significantly greater proportion of patients receiving infliximab demonstrated mucosal healing and were able to discontinue corticosteroids compared to patients receiving placebo (10). The efficacy of infliximab in hospitalized patients that have failed intravenous (IV) corticosteroids has thus far not been impressive and appears less effective than cyclosporine in this setting. However, there may be a role for those that are unable to take azathioprine or 6-mercaptopurine, a group in which cyclosporine would usually not be considered.
The currently approved and recommended dosing regimen for both CD and UC is a 5 mg/kg loading dose at weeks 0, 2, and 6, followed by repeat infusions every 8 weeks. The infusions can be given more frequently or the dose increased to 7.5 or 10 mg/kg, if needed. Current data suggest that intermittent therapy, also called ondemand therapy, with infliximab should be avoided in most situations.
Adalimumab
Adalimumab (Humira; Abbott Laboratories, Chicago, Illinois) is a fully human, subcutaneously (SQ) administered IgG1 monoclonal antibody to TNF-α, which like infliximab can induce T-cell apoptosis and cytotoxicity. Adalimumab was approved by the FDA for the treatment of CD in 2007, and trials of adalimumab in UC have been initiated. The bioavailability of adalimumab is estimated to be 64% in healthy volunteers, and it has a 2-week half-life (11). CLASSIC 1, a large double-blind, randomized controlled trial, demonstrated that adalimumab was effective in achieving clinical remission and response in patients with moderate-to-severe CD that were infliximab naïve. In this study, remission was achieved at week 4 in 24% (p = 0.06) and 36% (p = 0.001), with the two highest doses of adalimumab (80 mg/40 mg and 160 mg/80 mg at week 0/week 2) versus 12% for placebo (12).
Among patients that completed CLASSIC 1, 276 entered the CLASSIC 2 study. In CLASSIC 2, all patients received adalimumab 40 mg SQ at weeks 0 and 2, with those remaining in clinical remission at both weeks 0 and 4 re-randomized to receive either adalimumab 40 mg every other week (eow), adalimumab 40 mg/wk, or placebo for 1 year. Of the 55 patients that remained in clinical remission at both weeks 0 and 4, 79% of those that received adalimumab 40 mg eow and 83% of those that received adalimumab 40 mg/wk remained in clinical remission at week 56 compared to 44% of those on placebo. In addition, 204 patients that were not in clinical remission at both weeks 0 and 4 entered an open-label cohort in which they received adalimumab 40 mg SQ eow for 1 year, with the ability to dose escalate to 40 mg SQ per week for flares or persistent lack of response. Among the open-label patients, 46% remained in remission after 1 year and 65% achieved a 100-point reduction in Crohn’s Disease Activity Index (CDAI) (13).
More recently, the results of two Phase 3 double-blind, placebo-controlled trials (CHARM and GAIN) have been reported. The CHARM trial used a design similar to ACCENT I, in which 499 of 854 patients (58%) with moderate-to-severe CD who responded to open-label induction therapy with adalimumab were randomized to receive 1 of 3 therapies: adalimumab 40 mg eow, adalimumab 40 mg/wk, or placebo. The rates of clinical remission at both weeks 26 and 56 were significantly higher for patients treated with adalimumab (regardless of dose) compared to placebo. At week 56, the rates of clinical remission were 12%, 36%, and 41% for placebo, adalimumab eow, and adalimumab per week, respectively (14). Subanalysis of the CHARM data demonstrated that adalimumab is also significantly superior to placebo in maintaining steroid-free remission (off steroids for >90 days) and in maintaining complete fistula remission at weeks 26 and 56 (14).
The GAIN trial revealed that adalimumab was efficacious in inducing clinical response and remission in patients that previously lost response or developed side effects to infliximab (secondary failure). In this study, 325 patients with
moderate-to-severe CD with secondary failure to infliximab were randomized to receive adalimumab 160 mg at week 0 and 80 mg at week 2 versus placebo. The clinical remission rate at week 4 was 21% for the adalimumab-treated group versus 7% for the placebo-treated group (p <0.001). The improved remission rates in the adalimumab-treated group were significant, irrespective of whether patients developed intolerance to or lost response to infliximab and independent of the presence of anti-infliximab antibodies (15).
moderate-to-severe CD with secondary failure to infliximab were randomized to receive adalimumab 160 mg at week 0 and 80 mg at week 2 versus placebo. The clinical remission rate at week 4 was 21% for the adalimumab-treated group versus 7% for the placebo-treated group (p <0.001). The improved remission rates in the adalimumab-treated group were significant, irrespective of whether patients developed intolerance to or lost response to infliximab and independent of the presence of anti-infliximab antibodies (15).
The approved dosing regimen for adalimumab for CD is 160 mg subcutaneously at week 0 followed by 80 mg at week 2, and then 40 mg eow. The dose can be escalated to 40 mg every week or 80 mg eow if needed.
Certolizumab Pegol
Certolizumab pegol (Cimzia; UCB, Smyrna, Georgia) is a humanized monoclonal anti-TNF-α antibody Fab fragment (95% human, 5% murine) linked to polyethylene glycol that is administered by subcutaneous injection. The addition of polyethylene glycol extends the plasma half-life of the molecule and thus reduces the dosing frequency (16). The half-life of certolizumab pegol is approximately 14 days, with an 80% bioavailability. Unlike infliximab and adalimumab, certolizumab pegol lacks an Fc region and therefore does not fix complement or induce antibody-mediated cytotoxicity. Certolizumab pegol is approved for reducing the signs and symptoms of CD and maintaining clinical response in adult patients with moderate-to-severe active disease who have an inadequate response to conventional therapy.
Two Phase 3 double-blind, placebo-controlled trials (PRECiSE 1 and PRECiSE 2) have shown evidence that certolizumab pegol is effective in the induction and maintenance of response in CD. In PRECiSE 1,662 patients with moderate-to-severe CD were randomized to receive placebo or certolizumab pegol 400 mg at weeks 0, 2, and 4, and then every 4 weeks until week 24. The combined clinical response rate at weeks 6 and 26 was 21.5% for certolizumab pegol and 12.3% for placebo (p <0.05) (17).
PRECiSE 2 followed a design similar to that of ACCENT I for infliximab and CHARM for adalimumab. In PRECiSE 2, 428 of 668 (64%) patients who responded to open-labeled induction therapy with certolizumab pegol were randomized to receive certolizumab pegol 400 mg or placebo every 4 weeks through week 24. Clinical response at week 26 was maintained by 63% of patients in the certolizumab pegol-treated group versus 36% of patients in the placebo-treated group (p <0.001), and clinical remission rates were 48% and 29% for certolizumab pegol and placebo, respectively (p <0.001) (18).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


