Over the last 2 decades the evolution of alpha-blockers for lower urinary tract symptoms (LUTS)/benign prostatic hyperplasia (BPH) has been to preserve effectiveness, improve tolerability, and eliminate dose titration. Today, alpha-blockers represent the first-line treatment of most men with BPH whereby the primary objective is relief from bothersome LUTS.
Key points
- •
Over the last 2 decades the evolution of alpha-blockers for lower urinary tract symptoms (LUTS)/benign prostatic hyperplasia (BPH) has been to preserve effectiveness, improve tolerability, and eliminate dose titration.
- •
Today, alpha-blockers represent the first-line treatment of most men with BPH whereby the primary objective is relief from bothersome LUTS.
- •
The thought that alpha blockers only improve LUTS by relieving BOO is likely an oversimplification.
The first selective alpha-blocker was approved for the treatment of lower urinary tract symptoms (LUTS)/benign prostatic hyperplasia (BPH) in 1992. The evolution of alpha-blockers for LUTS/BPH has been to preserve effectiveness and improve tolerability following administration of a single daily dose without requirement for dose titration. Today, alpha-blockers represent the first-line treatment of most men with BPH whereby the primary objective is relief from bothersome LUTS. The proposed mechanism of action, which is to decrease bladder outlet obstruction (BOO) via smooth muscle relaxation, is an oversimplification.
Historical perspective
The evolution of alpha-blockers for the treatment of BPH represents one of the first triumphs of translational medicine.
In 1975, Caine and colleagues investigated the in vitro smooth muscle contractile properties of tissue strips derived from human prostates. Human prostate tissue was shown to elicit a strong contractile response in the presence of phenylephrine, a potent alpha agonist. At the time, the adrenergic receptors were subclassified simply as alpha and beta. The phenylephrine-induced contraction was inhibited by phenoxybenzamine, a selective inhibitor of alpha adrenoceptors. Based on these experiments, Caine and associates proposed that the pathophysiology of BPH was mediated in part by prostate smooth muscle tension and speculated that the disease would be effectively treated by alpha-blockade.
In 1975 Caine and colleagues published the first study suggesting alpha-blockers were effective for the treatment of BPH. A subsequent randomized placebo-controlled study confirmed that phenoxybenzamine improved both uroflow rates and prostatism, which at the time was the terminology describing LUTS arising from the enlarged prostate. The primary limitation of phenoxybenzamine was the adverse events, including high rates of tiredness, dizziness, impaired ejaculation, nasal stuffiness, and hypotension. The use of phenoxybenzamine never gained widespread use for treating BPH.
Historical perspective
The evolution of alpha-blockers for the treatment of BPH represents one of the first triumphs of translational medicine.
In 1975, Caine and colleagues investigated the in vitro smooth muscle contractile properties of tissue strips derived from human prostates. Human prostate tissue was shown to elicit a strong contractile response in the presence of phenylephrine, a potent alpha agonist. At the time, the adrenergic receptors were subclassified simply as alpha and beta. The phenylephrine-induced contraction was inhibited by phenoxybenzamine, a selective inhibitor of alpha adrenoceptors. Based on these experiments, Caine and associates proposed that the pathophysiology of BPH was mediated in part by prostate smooth muscle tension and speculated that the disease would be effectively treated by alpha-blockade.
In 1975 Caine and colleagues published the first study suggesting alpha-blockers were effective for the treatment of BPH. A subsequent randomized placebo-controlled study confirmed that phenoxybenzamine improved both uroflow rates and prostatism, which at the time was the terminology describing LUTS arising from the enlarged prostate. The primary limitation of phenoxybenzamine was the adverse events, including high rates of tiredness, dizziness, impaired ejaculation, nasal stuffiness, and hypotension. The use of phenoxybenzamine never gained widespread use for treating BPH.
Development of selective alpha1 blockers for benign prostatic hyperplasia
In the early 1980s, the alpha adrenoceptors were subclassified into alpha 1 and alpha 2. Lepor and Shapiro were first to characterize both the alpha 1 and alpha 2 adrenoceptors in human prostate tissue using radio-ligand binding studies and subsequently observed that the contractile properties of prostate smooth muscle was mediated primarily by the alpha 1 subtype.
Prazosin was one of the first alpha 1 blockers developed for the treatment of hypertension. The first multicenter randomized placebo-controlled trial of any selective alpha 1 blocker for symptomatic BPH was reported my Kirby and colleagues. Prazosin significantly improved both the severity of prostatism and peak flow rate (PFR) compared with placebo.
The primary advantage of prazosin over phenoxybenzamine was better tolerance presumably by eliminating the adverse events mediated by alpha 2 adrenoceptor blockade. Although a legitimate comparative trial between prazosin and phenoxybenzamine was never performed, the effectiveness of the two drugs seems to be comparable. Like phenoxybenzamine, the limitation of prazosin was the first dose effect, which required a dose titration to an effective multiple daily dose. No effort was made to seek Food and Drug Administration (FDA) approval for prazosin for the indication of symptomatic BPH presumably because of the limited patent life and the lack of general interest in developing and bringing to market medical therapy for BPH.
The next major advance in the development of alpha-blockers for the treatment of symptomatic BPH was the availability of long-acting selective alpha 1 blockers, such as terazosin and doxazosin. Both drugs were approved by the FDA for the treatment of hypertension in the 1980s. The longer half-life of both drugs allowed for once-daily dosing.
Terazosin was the first of the long-acting selective alpha 1 blockers to be approved by the FDA for the treatment of symptomatic BPH in 1992. Three randomized multicenter placebo-controlled studies leading to the approval of terazosin showed significant treatment-related decreases in symptom severity and increases in PFR over placebo ( Fig. 1 ). The author served as the national principal investigator for the first randomized study comparing placebo, 2, 5, and 10 mg of terazosin. Because of the potential safety concerns associated with administering an antihypertensive drug to normotensive men, all subjects were admitted to a monitored hospital facility for 72 hours. The requirement for 3-day hospital observation greatly limited patient accrual. Eventually, the FDA relaxed the in-hospital monitoring requirement to 24 hours and the study was completed. The titration to fixed-dose study was not powered to show whether differences between the doses were statistically significant, although there was a suggestion that improvement in LUTS was dose dependent. Overall, terazosin was well tolerated. Asthenia/fatigue, postural hypotension, dizziness, and somnolence were the treatment-related adverse events ( Table 1 ).

| Adverse Effect | Terazosin (%) (n = 636) | Placebo (%) (n = 360) |
|---|---|---|
| Asthenia/fatigue | 7.4 a | 3.3 |
| Postural | 3.9 a | 0.8 |
| Hypotension | ||
| Dizziness | 9.1 a | 4.2 |
| Somnolence | 3.6 a | 1.9 |
| Nasal congestion/rhinitis | 1.9 a | 0 |
| Impotence | 1.6 a | 0.6 |
The FDA required a long-term extension study in order to demonstrate the durability of efficacy. Subjects enrolled in the pivotal randomized placebo-controlled studies were given the opportunity to participate in a long-term open-label study that demonstrated durability of efficacy and emergence of no safety issues.
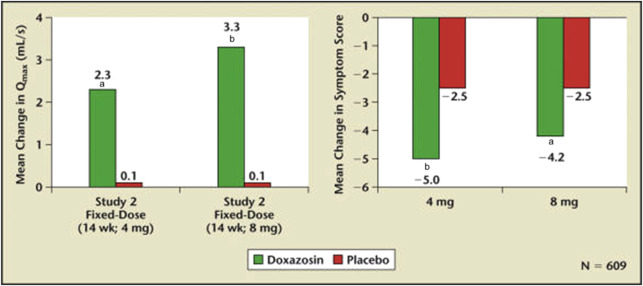
The chemical structure of terazosin and doxazosin are quite similar. It is not surprising that multicenter randomized placebo-controlled FDA registration studies for doxazosin and terazosin were virtually identical as far as treatment-related efficacy and side effects. The study designs included a titration to a fixed dose with the dose progressively increasing from 2 mg to 4 mg to 8 mg. The efficacy of these trials comparing placebo, 4 mg, and 8 mg of doxazosin is shown in Fig. 2 . In the titration to fixed dose study, a dose response to doxazosin was not observed. The side effects attributable to doxazosin were fatigue and dizziness ( Table 2 ). The FDA approved doxazosin for the treatment of BPH in the mid 1990s.
| Adverse Effect | Doxazosin (%) (n = 665) | Placebo (%) (n = 300) |
|---|---|---|
| Dizziness (includes vertigo) | 15.6 a | 9.0 |
| Fatigue | 8.0 a | 1.7 |
| Hypotension | 1.7 a | 0 |
| Edema | 2.7 a | 0.7 |
| Dyspnea | 2.6 a | 0.3 |
A long-term open-label study of doxazosin showed durability of efficacy and no emerging new safety issues.
In clinical practice, it is reasonable to assume that nonresponders will choose not to stay on the drug. Therefore, the performance of a drug in randomized placebo-controlled trials underestimates the effectiveness of the drug in clinical practice. MacDiarmid and colleagues provide the most compelling evidence for the dose response to alpha-blockers in clinical practice. Responders to 4 mg daily doxazosin were randomized to remain on 4 mg or to increase the dose to 8 mg. Very significant improvements in both symptom scores and PFR were observed between the 4- and 8-mg doses. This study supported the author’s recommendation to titrate both terazosin and doxazosin to the maximal tolerable dose. One of the more troublesome side effects leading to withdrawal of both terazosin and doxazosin was asthenia and dizziness. In order to improve tolerability, a second recommendation was to administer the doses at bedtime.
Tamsulosin was the next alpha 1 blocker to be FDA approved in the United States. Both pivotal registration trials compared daily placebo versus 4 mg and 8 mg tamsulosin. The initial dose for all men randomized to tamsulosin was 4 mg with a subsequent titration to the 8-mg dose. Both doses of tamsulosin were superior to placebo at relieving symptoms and increasing PFRs ( Fig. 3 ). Although the study was not powered to show differences between the 4- and 8-mg doses, in one of the pivotal trials the 8-mg dose was significantly superior to the 4-mg dose for improving LUTS. The adverse events attributable to tamsulosin were dose dependent and included dizziness, asthenia, and ejaculatory dysfunction. The incidences of ejaculatory dysfunction for placebo, 4 mg, and 8 mg tamsulosin were 0.2%, 8.4%, and 18.1%, respectively ( Table 3 ). Interestingly, ejaculatory dysfunction was not previously observed with terazosin or doxazosin. Initially the pathophysiology of ejaculatory dysfunction was attributed to retrograde ejaculation due to alpha 1 blockade of bladder neck smooth muscle. Wolters and Hellstron demonstrated that the tamsulosin ejaculatory dysfunction was anejaculation due to alpha-mediated inhibition of vassal and seminal vesicle smooth muscle.

| Adverse Effect | Tamsulosin 0.4 mg (%) (n = 502) | Tamsulosin 0.8 mg (%) (n = 492) | Placebo (%) (n = 493) |
|---|---|---|---|
| Dizziness | 14.9 | 17.1 | 10.1 |
| Abnormal ejaculation | 8.4 | 18.1 | 0.2 |
| Asthenia | 7.8 | 8.5 | 5.5 |
| Libido decreased | 1.0 | 2.0 | 1.2 |
| Amblyopia | 0.2 | 2.0 | 0.4 |
A long-term open-label extension study demonstrated both the durability of response and the emergence of no safety issues.
The advantage of tamsulosin over terazosin and doxazosin was the ability to administer a therapeutic dose without requirement for dose titration and the lack of postural hypotension. Initially, the pharmacologic property of tamsulosin that eliminated the requirement for dose titration was its presumed alpha 1a subtype selectivity. However, the binding and functional studies demonstrated that tamsulosin exhibited only a 10-fold selectivity for the alpha 1a versus alpha 1b subtypes and no alpha 1a versus alpha 1d selectivity. This level of selectivity is likely not clinically relevant. In retrospect, the pharmacologic advantage of eliminating titration to an effective dose was explained by the slow-release formulation and not its negligible alpha 1 subtype selectivity.
There are several reasons why only the 4-mg dose was marketed for BPH. The manufacturers were able to claim that tamsulosin was the only alpha 1 blocker achieving a therapeutic response without dose titration. In addition, it was not feasible to manufacture an 8-mg single dose. There also seemed to be a significant dose response related to adverse events. Despite the high rates of ejaculatory dysfunction even at the 4-mg dose, tamsulosin became the market leader most likely because of the lack of dose titration and because generic terazosin and doxazosin were commercially available and the branded drugs were no longer aggressively promoted by the industry.
The initial formulation of alfuzosin approved in Europe required twice-daily dosing. A slow-release formulation of alfuzosin (alfuzosin SR) was subsequently developed, which allowed for once-daily dosing without the requirement for titration to an effective dose. Alfuzosin SR was the last alpha 1 blocker approved by the FDA for the treatment of BPH based on 2 pivotal randomized placebo-controlled studies. Despite the fact alfuzosin did not exhibit any alpha 1 subtype selectivity, it was marketed as a uro-selective drug because in vivo studies in the cat model suggested the drug was more potent for relieving prostate urethral pressures than altering blood pressure. The mechanism of physiologic uro-selectivity was attributed to enhanced penetration of the drug into the prostate. Subsequent in vivo studies failed to show physiologic uro-selectivity in the canine model. The two alfuzosin pivotal trials demonstrated both efficacy ( Fig. 4 ) and excellent tolerance ( Table 4 ). The primary advantage of alfuzosin was the lack of ejaculatory dysfunction associated with tamsulosin and lower incidences of asthenia and dizziness associated with terazosin and doxazosin. Because of the absence of ejaculatory dysfunction, alfuzosin was positioned for men who would be bothered by anejaculation. A comparison of the efficacy from all the pivotal trials suggests that alfuzosin is the least effective of the alpha 1 blockers. The superior tolerability and inferior efficacy suggests the 10-mg dose was achieving a relatively low level of alpha 1 blockade.

Related posts:
 The Epidemiology of Benign Prostatic Hyperplasia Associated with Lower Urinary Tract Symptoms
The Epidemiology of Benign Prostatic Hyperplasia Associated with Lower Urinary Tract Symptoms
 Diagnostic Work-Up of Lower Urinary Tract Symptoms
Diagnostic Work-Up of Lower Urinary Tract Symptoms
 Antimuscarinics, β-3 Agonists, and Phosphodiesterase Inhibitors in the Treatment of Male Lower Urinary Tract Symptoms
Bipolar, Monopolar, Photovaporization of the Prostate, or Holmium Laser Enucleation of the Prostate
Robotic-Assisted Simple Prostatectomy
Sexual Side Effects of Medical and Surgical Benign Prostatic Hyperplasia Treatments
Antimuscarinics, β-3 Agonists, and Phosphodiesterase Inhibitors in the Treatment of Male Lower Urinary Tract Symptoms
Bipolar, Monopolar, Photovaporization of the Prostate, or Holmium Laser Enucleation of the Prostate
Robotic-Assisted Simple Prostatectomy
Sexual Side Effects of Medical and Surgical Benign Prostatic Hyperplasia Treatments
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


