Fig. 59.1
a, b Xanthomas on the hands of a child with Alagille syndrome
The natural history of the liver disease in ALGS has a unique course. For those children with significant cholestasis in infancy, the hepatic involvement generally follows a more severe course in the first 5 years of life after which it appears to improve for most patients. This spontaneous improvement is poorly understood, but well documented. In approximately 10–20 %, the cholestasis persists unabated or progresses to end-stage liver disease (ESLD). For those children with mild cholestasis or hepatitis in early childhood, there is no progression of liver disease in later life. It is difficult to predict early on, which ALGS children with cholestasis in early childhood will eventually require liver transplantation and which will spontaneously improve. There are no known genotypic or radiologic predictors of liver disease progression in ALGS. A review of laboratory data of ALGS patients showed that bilirubin and cholesterol levels before the age of 5 may aid in distinguishing patients at high and low risk of problematic cholestasis in later childhood. More specifically, mean levels of total bilirubin (TB)> 6.5 mg/dL (111 μmol/L), conjugated bilirubin (CB)> 4.5 mg/dL (77 μmol/L), and cholesterol > 520 mg/dL (13.3 mmol/L) are strongly associated with severe liver disease in later life, whereas levels lower than this are associated with a good hepatic outcome [13]. These data may assist the clinician in predicting which children might go on to resolve their cholestasis, and thereby avoid unnecessary liver transplantation in young children with ALGS.
Of note, there have been several case reports of hepatocellular carcinoma in patients with ALGS, including as young as 4 years of age [14–16]. Most but not all patients who developed hepatocellular carcinoma also had progressed to cirrhosis.
Bile duct paucity is the hallmark histopathologic feature of ALGS. The normal bile duct to portal space ratio is between 0.9 and 1.8. Bile duct paucity is defined as a ratio of bile duct to portal tract that is less than 0.9. Bile duct paucity, however, is not present in infancy in many patients ultimately shown to have ALGS. Several studies of serial liver biopsies have demonstrated that paucity is more common later in infancy and childhood [4, 17, 18]. Emerick et al. found that paucity was present in 60 % of 48 infants younger than 6 months of age but in 95 % of 40 who underwent biopsy after 6 months [4].
Cardiac
In a comprehensive evaluation of 200 ALGS subjects, cardiovascular involvement was present in 94 % [19], with right-sided lesions being the most prevalent. Pulmonary artery anomalies are the most common abnormality identified (76 %) and may occur in isolation or in combination with structural intracardiac disease [19]. The most common congenital defect is tetralogy of Fallot (TOF), which occurs in 7–12 % [4, 19]. Approximately, 40 % of patients with ALGS demonstrating TOF have pulmonary atresia, representing a more severe phenotype. Cardiac disease accounts for nearly all of the early deaths in ALGS. Patients with intracardiac disease have approximately a 40 % rate of survival to 6 years of life, compared with a 95 % survival rate in patients with ALGS without intracardiac lesions [4].
Facial Features
A characteristic facial appearance is one of the most penetrant features of ALGS (for JAG1-associated disease). These features include a prominent forehead, deep-set eyes with moderate hypertelorism, a pointed chin, and a saddle or straight nose with a bulbous tip. The combination of these features gives the face a triangular appearance (Fig. 59.2). The facies may be present early in infancy but in general becomes more apparent with increasing age. In adults, the forehead is much less prominent, and the protruding chin is more noticeable so that the face loses the triangular appearance.
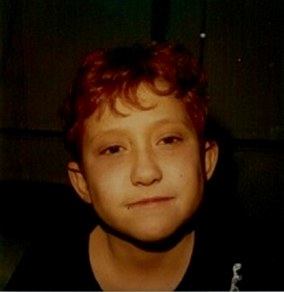
Fig. 59.2
Facial features of Alagille syndrome
It should be noted that among the few patients reported to date, there appears to be a lower penetrance of characteristic ALGS facial features in patients with NOTCH2 mutations, and it is therefore a less valuable diagnostic tool in this group [20].
Ophthalmologic
The ocular abnormalities of patients with ALGS do not generally affect vision but are important as diagnostic tools. A large and varied number of ocular abnormalities have been described, though posterior embryotoxon is the most important diagnostically. Posterior embryotoxon is a prominent, centrally positioned Schwalbe’s ring (or line) at the point at which the corneal endothelium and the uveal trabecular meshwork join and is visible on slit-lamp examination. Posterior embryotoxon occurs in 56–88 % of patients with ALGS and was also detected in 22 % of children evaluated in a general ophthalmology clinic [21]. Posterior embryotoxon is seen in other multisystem disorders such as chromosome 22q deletion, as well. The Axenfeld anomaly, seen in 13 % of patients with ALGS, is a prominent Schwalbe’s ring with attached iris strands and is associated with glaucoma. Optic disk drusen identified using ocular ultrasonography has been described in ALGS patients with high prevalence but this test is not routinely performed [22, 23].
Skeletal Involvement
The most characteristic skeletal finding is the sagittal cleft or butterfly vertebrae, which is found in 33–87 % of patients with ALGS [4, 24–26]. This relatively uncommon anomaly may occur in normal individuals and is also seen in other multisystem abnormalities, such as 22q deletion syndrome and vertebral defects, anal atresia, tracheoesophageal fistula, and radial and renal defect (VATER) syndrome. The affected vertebral bodies are split sagittally into paired hemivertebrae because of a failure of the fusion of the anterior arches of the vertebrae. Generally, these are asymptomatic and of no structural significance.
Other associated skeletal abnormalities include an abnormal narrowing of the adjusted interpedicular space in the lumbar spine, a pointed anterior process of C1, spina bifida occulta, fusion of the adjacent vertebrae, hemivertebrae, the absence of the 12th rib, and the presence of a bony connection between ribs. In addition, supernumerary digital flexion creases have been described in one third of patients [27].
Severe metabolic bone disease with osteoporosis and pathologic fractures is common in patients with ALGS. Recurrent fractures, particularly of the femur, have been cited as an indication for liver transplantation. Preliminary survey data suggest that there is a propensity towards pathologic lower extremity long bone fractures in ALGS [28]. A number of factors may contribute to osteopenia and fractures, including severe chronic malnutrition , vitamin D and vitamin K deficiency, chronic hepatic and renal disease, and magnesium deficiency. It is not yet known whether there is an intrinsic defect in cortical or trabecular structure of the bones in patients with ALGS. Olsen evaluated bone status in prepubertal children with ALGS and identified significant deficits in bone size and bone mass that were related to fat absorption but not dietary intake [29].
ALGS patients are frequently found to have short stature; this is likely multifactorial in origin resulting from cholestasis and malabsorption, congenital heart disease, and genetic predisposition. A validated growth curve for ALGS individuals is not yet available.
Renal Involvement
Renal involvement in ALGS has been widely reported on an individual case basis or as part of a larger report on general features of ALGS. The prevalence of renal involvement in larger series ranges from 40 to 70 % such that it has been proposed that renal anomalies now be considered a disease-defining criterion in ALGS. In a large retrospective study, there was a prevalence of 39 % of renal anomalies or disease. The most common renal involvement was renal dysplasia (58.9 %), followed by renal tubular acidosis (9.5 %), vesicoureteric reflux (8.2 %), and urinary obstruction (8.2 %) [30]. Hypertension in patients with ALGS can be of cardiac, vascular, or renal etiology.
Functional and structural evaluation of the kidneys should be undertaken in all patients. Renal function should be reassessed during the evaluation for liver transplantation.
Vascular Involvement
Unexplained intracranial bleeding is a recognized complication and cause of mortality in ALGS. Intracranial bleeds occur in approximately 15 % of patients; in 30–50 % of these events, the hemorrhage is fatal [4, 25]. There does not seem to be any pattern to the location and/or severity of the intracranial bleeding, which ranges from massive fatal events to asymptomatic cerebral infarcts. Epidural, subdural, subarachnoid, and intraparenchymal bleeding have been reported. The majority of this bleeding has occurred in the absence of significant coagulopathy or trauma. Lykavieris studied a cohort of 174 individuals with ALGS and identified 38 patients (22 %) who had 49 bleeding episodes [31]. All these hemorrhages occurred in the absence of liver failure, with normal median platelet counts and prothrombin times, suggesting that ALGS patients may be at particular risk for bleeding.
Underlying vessel abnormalities in the central nervous system that could explain the occurrence of bleeding and stroke in ALGS have been described in some of these patients [5, 25, 32]. Aneurysms of the basilar and middle cerebral arteries and various internal carotid artery anomalies have been described. Moyamoya disease (progressive intracranial arterial occlusive disease) also has been previously described in several children with ALGS. Emerick et al. prospectively studied 26 patients with ALGS using magnetic resonance imaging (MRI) with angiography of the head. Cerebrovascular abnormalities were detected in 10 of 26 patients (38 %). One hundred percent of symptomatic patients had detected abnormalities, and 23 % of screened, asymptomatic patients had detected anomalies [32]. These results suggest that MRI with angiography is useful in detecting these lesions and may have a valuable role in screening for treatable lesions such as aneurysms. The current recommendation is for all asymptomatic ALGS patients to have a screening MRI/magnetic resonance (MR) angiography as a baseline and for physicians to have a low threshold for reimaging ALGS patients in the event of any symptoms, head trauma, or suspicious neurologic signs and prior to major surgical interventions.
Systemic vascular abnormalities have also been well documented in ALGS. Aortic aneurysms and coarctations, renal artery, celiac artery, superior mesenteric artery, and subclavian artery anomalies have all been described. In the evaluation of a large cohort of ALGS patients, 9 % (25 of 268) with noncardiac vascular anomalies or events were identified [5]. In addition, vascular accidents accounted for 34 % of the mortality in this cohort. These findings suggest that vascular abnormalities have been under-recognized as a potentially devastating complication of ALGS.
Genetics of Alagille Syndrome
ALGS is inherited in an autosomal dominant manner, with highly variable expressivity. It is a genetically heterogeneous disorder and may be caused by mutations in either JAG1 (seen in 94 % of clinically defined probands) or NOTCH2 (seen in approximately 1 %) [33–35]. Jagged1 is a cell surface protein that serves as a ligand for the four Notch receptors (Notch1, 2, 3, and 4), and together these proteins begin the cascade of events that turn on the Notch signaling pathway. The Notch signaling pathway is involved in the determination of cell fate and as such plays a crucial role in normal development .
Gene Identification and Mutation Analysis
JAG1 was identified as the cause of ALGS in 1997 [33, 35]. To date, more than 430 JAG1 mutations have been identified in patients with ALGS. Utilizing current screening techniques, the mutation detection rate is 94 % [36]. The frequency of sporadic mutations (i.e., new in the proband) is approximately 60–70 %. Approximately, 75 % of ALGS patients have JAG1 protein-truncating (frameshift or nonsense or splice-site) mutations [8, 36–38]. Approximately, 7 % have gene deletions. Missense mutations are identified in 15 %. Haploinsufficiency, a decrease in the amount of the normal protein, is hypothesized to be the mechanism causing ALGS.
ALGS associated with NOTCH2 mutations was described in 2006. Thus far, ten patients with unique NOTCH2 mutations have been described [20, 34].
A small fraction (3–5 %) of ALGS individuals have deletions of chromosome 20p. Genome-wide single-nucleotide polymorphism (SNP) analysis of 25 patients with ALGS revealed 21 deletions ranging from 95 kb to 14.62 Mb [39]. Patients with deletions greater than a critical 5.4 Mb region had additional phenotypic features not usually associated with ALGS such as developmental delay and hearing loss.
Genotype–Phenotype Correlations
JAG1 Mutations
Although the ALGS phenotype is highly variable, there is no apparent correlation with JAG1 genotype in the majority of patients. A study of 53 JAG1 mutation-positive relatives of a cohort of ALGS probands demonstrated that only 53 % met the clinical criteria for a diagnosis of ALGS, including 11 of 53 with obvious clinical features that would easily have led to a diagnosis of ALGS and 17 of 53 (32 %) who had mild features that would have only been apparent on targeted evaluation following the diagnosis of a proband in their family (i.e., discovery of elevation of liver enzymes or posterior embryotoxon in an asymptomatic individual) [7]. This underscores the variable clinical consequences associated with a JAG1 mutation and suggests the presence of genetic modifiers.
NOTCH2 Mutations
Diagnostic Considerations
Clinical Diagnostics
The majority of infants with ALGS are evaluated for conjugated hyperbilirubinemia in the first weeks or months of life. ALGS is occasionally misdiagnosed as biliary atresia because of the overlap of biochemical, scintigraphic, histologic, and cholangiographic features. Serum bilirubin, bile acid, and GGT levels typically are elevated in both of these disorders. Ultrasound findings in both conditions may reveal small or apparently absent gallbladders. Excretion of nuclear tracer (diisopropylacetanilido iminodiacetic acid; DISIDA) into the duodenum excludes biliary atresia, but non-excretion of tracer is also possible in ALGS. There was no excretion of scintiscan in 61 % of 36 infants with ALGS [4].
Although a liver biopsy is not mandatory to diagnose ALGS, it remains an important step in differentiating between ALGS and biliary atresia. In biliary atresia, bile duct proliferation is the typical histologic lesion. In ALGS, paucity is evident in 60 % of infants younger than 6 months but in 95 % of older patients [4]. Unfortunately, there may be a normal number of ducts early in the course of biliary atresia and also in some patients with ALGS, and bile duct proliferation occasionally occurs in infants with ALGS. Giant cell hepatitis is also seen in both disorders. Finally, it should be noted that bile duct paucity, if present, is not diagnostic of ALGS and other diagnoses should be considered (e.g., alpha-1-antitrypsin deficiency, cystic fibrosis, cytomegalovirus infection, etc.).
An operative cholangiogram is the gold standard procedure to evaluate the extrahepatic and intrahepatic biliary tree; however, this can also be misleading in ALGS. The extra- and intrahepatic ducts are extremely small in patients with ALGS, and the cholangiogram commonly does not demonstrate communication proximally. In 37 % of 19 cholangiograms in infants with ALGS, there was no opacification of the proximal extrahepatic ducts, and, in another 37 %, the proximal extrahepatic tree was abnormally small [4]. The intrahepatic ducts were normal in only 10 % of 19 infants with ALGS, small or hypoplastic in 16 %, and not visualized in 74 %. Therefore, even the apparent gold standard test to differentiate ALGS and biliary atresia can be misleading.
Clinical features in extrahepatic organ systems may also help in the diagnostic evaluation. The list of abnormalities identified in the “major” organ systems and the list of other affected organs have grown appreciably. Thus, an echocardiogram, slit-lamp examination, renal ultrasound, and spinal X-ray are essential diagnostic tests when ALGS is suspected. It should be noted that several of the ALGS-defining features are present in normal individuals or other conditions. Heart murmurs are present in 6 % of all newborns, posterior embryotoxon appears in 22 % of the general population, and butterfly vertebrae are seen in 11 % of patients with 22q11 deletion. Furthermore, the facial features of ALGS patients are subtle during the first months of life making this an unreliable diagnostic tool in infancy.
With the advent of molecular testing for ALGS and the broader appreciation of the phenotypic variability, the diagnostic criteria for ALGS have been modified. To make a clinical diagnosis for an index case (proband) in the family, the original Alagille criteria hold, modified only so as no longer to require histology. Thus ALGS can be diagnosed clinically on the basis of cholestasis with at least three features from the list of characteristic Alagille facies, consistent cardiac disease, posterior embryotoxon, butterfly vertebrae, typical ALGS renal disease, and a structural vascular anomaly. In families with one definite clinically defined proband, other members with only two features should be considered as having ALGS.
Molecular Diagnostics
Molecular sequencing is now widely commercially available for JAG1 and NOTCH2 on a limited basis. An evaluation by fluorescence in situ hybridization (FISH) for deletions including the JAG1 gene will identify these deletions in less than 7 % of patients. A molecular diagnosis can assist in an atypical ALGS case and is also useful for genetic counseling and prenatal diagnosis. JAG1 sequencing identifies mutations in individuals with clinically defined ALGS in the majority of cases ( > 90 %). Individuals that have clinical features of ALGS but are not found to be carrying JAG1 mutations should have sequence analysis of NOTCH2 [34].
Once a JAG1 mutation is identified in a proband, it is simple to test parents and other relatives for the identified mutation. Mutations are inherited from an affected parent in 30–50 % of patients, whereas the mutations appear de novo in 60–70 % [8, 37]. If a parental mutation is identified, there is a 50 % risk for each future offspring to inherit the JAG1 mutation. However, it should be emphasized that expressivity of the disorder is highly variable, and it is not currently possible to predict disease severity. If no parental mutation is identified, then the recurrence risk is limited to the chance of germ-line mosaicism, which for multiple different disorders is estimated at from 1 to 3 %.
Prenatal genetic testing for ALGS is possible if a parental mutation has been identified. This requires amniocentesis or chorionic villous sampling and assessment for a known JAG1 mutation. Pre-implantation genetic diagnosis has also been successfully performed in ALGS. It is imperative to carefully counsel parents undergoing any type of prenatal testing since there are no genotype–phenotype correlations in ALGS, so it is not possible to make predictions about a child’s clinical course based on the type or presence of a mutation.
Management of Alagille Syndrome
Cholestasis Management
Patients with ALGS present significant management challenges due to profound cholestasis and complex multisystem disease [40, 41] . A sequential and additive approach to medical cholestasis therapy in ALGS is most appropriate. The most commonly used agents are listed in Table 59.1 with common side effects and described below .
Table 59.1
Medications for cholestasis in Alagille syndrome
Medication | Dose | Most frequent side effects |
|---|---|---|
Choleretics | ||
Ursodiol (Actigall) | 10–30 mg/kg/day, divided in 2 doses < div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |





