Provision of information above and beyond serum creatinine and/or urine output
Non-invasive test using easily accessible samples
Results rapidly available
Specific cut-off values to distinguish between normal and abnormal renal function
Ability to differentiate between AKI and chronic kidney disease
Ability to differentiate between intrinsic AKI and pre-renal fluid responsive azotemia
Reliability in the setting of common comorbidities
Correlation with severity of AKI
Prognostication of important outcomes (i.e. need for renal replacement therapy, mortality)
Differentiation between different aetiologies of AKI
Indication of duration of AKI
Tool to guide clinical management and allow monitoring
9.2 Types of Biomarkers
Biomarkers of AKI vary in their origin, function, distribution and time of release following renal injury (Table 9.2 and Fig. 9.1). They can be broadly divided into:
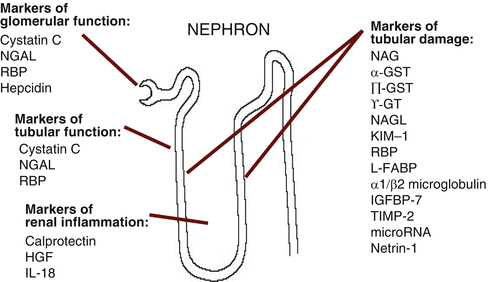
Table 9.2
AKI biomarkers in human studies
AKI biomarker | Production/origin | Handling by the kidney | Detection time after renal injury | Confounding factors |
|---|---|---|---|---|
Alanine aminopeptidase (AAP) Alkaline phosphatase (ALP) γ-glutamyl transpeptidase (γ-GT) | Enzymes located on the brush border villi of the proximal tubular cells | Released from brush border after damage to proximal tubular cells | Within <12 h | |
Calprotectin (activator of the innate immune system) | Cytosolic calcium-binding complex of two proteins of the S100 group (S100A8/S100A9) and derived from neutrophils and monocytes | Measure of local inflammatory activity; detectable in urine following intrinsic AKI | Within <12 h | Inflammatory bowel disease Urinary tract infection Probably CKD |
Cystatin C | 13 kDa cysteine protease inhibitor produced by all nucleated human cells and released into plasma at constant rate | Freely filtered in glomeruli and completely reabsorbed and catabolized by proximal tubular cells; no tubular secretion | 12–24 h post-renal injury | Systemic inflammation Malignancy Thyroid disorders Glucocorticoid disorder Smoking Hyperbilirubinaemia Hypertriglyceridaemia |
α glutathione S-transferase (α GST) | 47–51 kDa cytoplasmic enzyme produced in proximal tubule | Released into urine following tubular injury | Within 12 h | |
п glutathione S-transferase (п GST) | 47–51 kDa cytoplasmic enzyme produced in distal tubules | Released into urine following tubular injury | Within 12 h | |
Hepatocyte growth factor (HGF) | Antifibrotic cytokine produced by mesenchymal cells and involved in renal tubular cell regeneration after AKI | Within <12 h | ||
Hepcidin | 2.78 kDa peptide hormone produced in hepatocytes and other tissues; renoprotective role during ischaemia/reperfusion injury | Freely filtered with significant tubular uptake and catabolism (fractional excretion 2 %); higher levels in patients without AKI | Within 12–24 h | Systemic inflammation |
Insulin-like growth factor binding protein-7 (IGFBP-7) and Tissue metalloproteinase-2 (TIMP-2) | Metalloproteinases involved in cell cycle arrest | Released into urine after tubular epithelial injury | Within 12 h | |
Interleukin-18 (IL-18) | 18 kDa proinflammatory cytokine | Released into urine from proximal tubular cells following injury | 6–24 h after renal injury | Inflammation Sepsis Heart failure |
Kidney Injury Molecule–1 (KIM-1) | Transmembrane glycoprotein produced by proximal tubular cells after ischaemic or nephrotoxic injury | Released into urine following ischaemic or nephrotoxic tubular damage | 12–24 h after renal injury | Renal cell carcinoma Chronic proteinuria Chronic kidney disease Sickle cell nephropathy |
Liver-type fatty acid-binding protein (L-FABP) | 14 kDa intracellular lipid chaperone produced in proximal tubular cells and hepatocytes | Freely filtered in glomeruli and reabsorbed in proximal tubular cells; increased urinary excretion after tubular cell damage | 1 h after ischaemic tubular injury | Chronic kidney disease Polycystic kidney disease Liver disease Sepsis |
MicroRNA | Endogenous single-stranded molecules of non-coding nucleotides | Upregulated following tubular injury and detectable in plasma and urine | Within <20 h post-renal insult | Sepsis |
Monocyte chemoattractant peptide-1 (MCP-1) | Peptide expressed in renal mesangial cells and podocytes | Released into urine | ? | Variety of primary renal diseases |
N-acetyl-β-D-glucosaminidase (NAG) | >130 kDa lysosomal enzyme; produced in proximal and distal tubular cells (and non-renal cells) | Too large to undergo glomerular filtration; released into urine after tubular damage | 12 h | Diabetic nephropathy |
Neutrophil gelatinase-associated lipocalin (NGAL) also known as oncogene 24p3 | 25 kDa glycoprotein produced by epithelial tissues throughout the body | Plasma NGAL is excreted via glomerular filtration and undergoes complete reabsorption in healthy tubular cells NGAL is also produced in distal tubular segments and released into urine following tubular damage | Within 2–4 h | Sepsis Malignancy Chronic kidney disease Pancreatitis COPD Endometrial hyperplasia |
Netrin-1 | Laminin-related molecule, minimally expressed in proximal tubular epithelial cells of normal kidneys | Highly expressed in injured proximal tubules | Within 2–6 h | |
Retinol binding protein (RBP) | 21 kDa single-chain glycoprotein; specific carrier for retinol in the blood (delivers retinol from the liver to peripheral tissues) | Totally filtered by the glomeruli and reabsorbed but not secreted by proximal tubules; minor decrease in tubular function leads to excretion of RBP in urine | Within 12 h | Type II diabetes Obesity Acute critical illness |
Fig. 9.1
Origin and function of novel AKI biomarkers (Modified from Ref. [3]). Abbreviations: AKI acute kidney injury, NGAL neutrophil gelatinase-associated lipocalin, NAG N-acetyl-β-D-glucosaminidase, GST glutathione S-transferase, γ-GT γ-glutamyl transpeptidase, KIM-1 Kidney Injury Molecule-1, IL-18 interleukin 18, RBP retinol binding protein, L-FABP liver-type fatty acid-binding protein, IGFBP-7 insulin-like growth factor binding protein-7, TIMP-2 tissue metalloproteinase–3, HGF hepatocyte growth factor
(a)
Markers of glomerular function: small molecular weight proteins that are present in the systemic circulation and undergo glomerular filtration (i.e. serum creatinine, cystatin C)
(b)
Markers of tubular function: molecules that are filtered and undergo tubular reabsorption (i.e. retinol-binding protein)
(c)
Markers of tubular injury, damage or repair: molecules that are released as a result of direct renal cell damage, inflammatory activation or following gene upregulation [i.e. Kidney Injury Molecule 1 (KIM-1) or Interleukin 18 (IL 18)]
Biomarkers of kidney damage (NGAL, KIM-1 or IL 18) can be utilized to describe the nature, severity and site of renal injury. They may also provide information related to the underlying pathogenesis and prognosis. In contrast, functional biomarkers (i.e. creatinine, cystatin C) represent changes in renal function independent of site of damage. Most biomarkers are either damage or functional markers but some fulfil both roles (i.e. NGAL).
In theory, these new biomarkers have great potential, especially when used in combination and measured sequentially. They have been studied in adult and paediatric patients with and without co-morbidities and in various clinical scenarios [Intensive Care Unit (ICU), emergency department, post-contrast exposure, following transplantation and after cardiac surgery]. Some studies were performed in well-defined settings where the exact timing of renal injury was known (i.e. after surgery), whereas others were undertaken in patient cohorts with a less defined onset of AKI, for instance in patients with sepsis. These differences account for some of the discrepant findings.
9.3 Novel AKI Biomarkers in Clinical Practice
9.3.1 Diagnosis of Early AKI
Although the risk factors for AKI are well known, the early diagnosis of AKI in high-risk patients remains a challenge. The most commonly encountered comorbidities associated with AKI are age, diabetes, hypertension, obesity, liver disease, congestive heart failure, vascular disease and chronic kidney disease (CKD), and the most common renal insults include sepsis, hypotension, nephrotoxic agents and cardiopulmonary bypass surgery [4].
Following a definite renal injury, serum creatinine rise lags by 24–36 h. As a result, the early stage of AKI often remains unnoticed. Many studies have focussed on the ability of biomarkers to diagnose AKI before a detectable serum creatinine rise in different clinical settings.
9.3.1.1 Subclinical AKI
Recent studies identified a unique cohort of patients with a transient elevation in urinary and plasma NGAL levels without detectable changes in serum creatinine [5, 6]. Affected patients had a greater risk of complications, a longer stay in ICU and a higher risk of dying compared to patients without elevated NGAL levels. These results imply the existence of a state of “subclinical AKI” where renal injury has occurred but glomerular function is still preserved. Whether this phase of AKI represents a golden window for effective therapeutic interventions will need to be investigated in future studies.
9.3.1.2 In the Emergency Department
The identification of patients with early AKI at a time when serum creatinine is still in the normal range may be particularly useful in patients presenting to the emergency department. However, existing data are conflicting. A study in emergency patients with suspected sepsis showed that a plasma NGAL (pNGAL) >150 ng/ml had a sensitivity of >80 % for predicting AKI but specificity was poor at 51 % [7].
A different study was performed in 635 patients who were admitted to hospital from the emergency department. It concluded that a single measurement of urinary NGAL (uNGAL) helped to distinguish acute renal injury from normal function, prerenal azotemia and CKD and was also highly predictive of clinical outcomes, including nephrology consultation, need for renal replacement therapy (RRT) and admission to the ICU [8]. However, the mean serum creatinine of those with AKI was already elevated at 495 μmol/L (standard deviation 486) at presentation in the emergency department.
A study in 207 consecutive patients presenting to the emergency department with acute heart failure demonstrated that after control for pre-existing chronic cardiac or kidney disease, serum creatinine but not pNGAL was an independent predictor of AKI [9]. In contrast, a multi-centre study in 665 patients admitted to hospital from the emergency department showed that adding serial pNGAL results to clinical judgement improved the prediction of AKI [10]. Results of further studies are awaited to decide how best to utilise novel AKI biomarkers in the emergency setting.
9.3.1.3 Post-cardiac Surgery
The most studied AKI biomarkers after cardiac surgery are those that reflect an inflammatory process (such as IL-18) or markers which are released by tubular cells following renal injury (such as NGAL and KIM-1). Studies have focussed on the ability to diagnose early AKI and to predict outcomes, including progression to more severe AKI, need for RRT and mortality [11–14]. The majority of studies concluded that NGAL, IL-18, cystatin C, KIM-1 and liver-type fatty acid-binding protein (L-FABP) indicated AKI earlier than serum creatinine. For instance, urine IL-18 and urine and plasma NGAL peaked within 6 h after admission to ICU which was well before a serum creatinine rise at 24–72 h [15]. In a different study, the addition of urine IL-18 and pNGAL results to a clinical risk model based on age, gender, ethnicity, diabetes, hypertension, preoperative renal function and cardio-pulmonary bypass time increased the area under the curve to predict AKI from 0.69 to 0.76 and 0.75, respectively [14].
Other studies focussed on the performance of new AKI biomarkers as indicators of severity and progression of renal injury. Measurement of 32 different biomarkers in 95 patients with AKI stage 1 after cardiac surgery showed that IL-18 was the best predictor for worsening AKI or death, followed by L-FABP, NGAL and KIM-1 [12]. A different study showed that п glutathione S-transferase (п GST) was best at predicting the progression to AKI stage 3 in patients with a raised serum creatinine after cardiac surgery, followed by NGAL, cystatin C, hepatocyte growth factor and KIM-1 [13]. Of note, IL-18 was not measured. Markers of cell cycle arrest have also shown promising results [16]. In high-risk patients after cardiac surgery, serial levels of urinary tissue inhibitor of metalloproteinases-2 (TIMP-2) and insulin-like growth factor-binding protein 7 (IGFBP7) performed well in predicting early AKI and also renal recovery.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


