The evaluation and management of neonates with ambiguous genitalia requires sensitivity, efficiency, and accuracy. The approach to these neonates is facilitated by a multidisciplinary team including urology, endocrinology, genetics, and psychiatry or psychology. Disorders of sex development (DSD) encompass chromosomal DSD, 46,XX DSD, and 46,XY DSD. The 46,XX DSD is the most common DSD and in the majority of these children congenital adrenal hyperplasia is the underlying etiology. The 46,XY DSD is a heterogeneous disorder that often results from a disruption in the production or response to testosterone, dihydrotestosterone, or Mullerian inhibitory substance. Chromosomal DSD includes conditions resulting from abnormal meiosis, including Klinefelter syndrome (47, XXY) and Turner syndrome. The evaluation of children with DSD demands a thorough physical examination, medical history, karyotype, metabolic panel, 17-OH progesterone, testosterone, luteinizing hormone, follicle stimulation hormone, and urinalysis. A radiographic evaluation should begin with an abdominal and pelvic ultrasound but may include magnetic resonance imaging, endoscopy, or laparoscopy.
Chapter
The evaluation and management of a newborn with ambiguous genitalia must be undertaken with immediacy and great sensitivity. The pediatric urologist, endocrinologist, geneticist, and child psychiatrist or psychologist should work closely with the family in pursuing a dual goal: to establish the correct diagnosis of the abnormality and, with input from the parents, determine gender based on the karyotype, endocrine function, and anatomy of the child. In this section the authors outline a practical approach to the neonate born with a disorder of sex development (DSD).
Nomenclature
Genital ambiguity in the neonate has been described for centuries and evidence for disorders of sexual differentiation exists from many ancient civilizations. The actual incidence of DSD is difficult to accurately determine because of the heterogeneity of the clinical presentation and the varied etiologies. Using birth registries, some authors have attempted to estimate the incidence of ambiguous genitalia at birth; The estimated incidence of clinically detectable ambiguous genitalia at birth in Germany is 2.2 per 10,000 births. Congenital adrenal hyperplasia is estimated to occur in approximately 1 per 16,000 births. Historically, the term intersex was used to characterize DSD and subcategories included male pseudohermaphrodite, female pseudohermaphrodite, and true hermaphrodite. These terms used gender in the nomenclature and were often considered controversial or disparaging. Therefore a revised nomenclature was proposed that incorporated genetic etiology and descriptive terminology while removing gender references. The main categories include sex chromosome DSD, 46,XX DSD, and 46,XY DSD. Some conditions can be placed into more than one category. Additionally, although the majority of infants with 46, XX DSD will be diagnosed with congenital adrenal hyperplasia (CAH), only approximately 50% of children with 46, XY DSD will have a definitive clinical diagnosis.
Diagnosis
Chromosomal sex is established at fertilization and the undifferentiated gonads subsequently develop into either testes or ovaries. A child’s phenotypic sex results from the differentiation of internal ducts and external genitalia under the influence of hormones and transcription factors. Any discordance among these processes results in ambiguous genitalia or DSD. Currently, the main categories of DSD are 46,XX DSD, 46,XY DSD, sex chromosome DSD, ovotesticular DSD, and 46,XX testicular DSD.
46, XX disorder of sex development
Girls with 46,XX DSD, the most common DSD, are 46,XX with normal ovaries and Müllerian derivatives. The sexual ambiguity is limited to masculinization of the external genitalia that occurs as a result of exposure to androgens in utero. Congenital adrenal hyperplasia (CAH), which accounts for the majority of patients with 46,XX DSD, describes a group of autosomal recessive disorders that arises from a deficiency in one of five genes required for the synthesis of cortisol ( Fig. 1 ). These five genes and the enzymes they encode are include CYP21 : 21-hydroxylase; CYP11 : 11β-hydroxylase, 18-hydroxylase and 18-oxidase; CYP 17 : 17α-hydroxylase and 17,20 lyase; 3β2HSD : 3β–hydroxysteroid dehydrogenase; and StAR : side chain cleavage enzyme. Although these biochemical defects are characterized by impaired cortisol secretion, only deficiencies in CYP21 and CYP11 are predominantly masculinizing disorders, and 3β2HSD to a lesser extent. Although the female fetus is masculinized because of overproduction of adrenal androgens and precursors, the affected boys have no genital abnormalities. In contrast, 3β2HSD , CYP17, and StAR deficiencies block cortisol synthesis and gonadal steroid production. Thus, boys have varying degrees of undermasculinization, whereas girls generally have normal external genitalia. CAH resulting in genital ambiguity in boys is discussed in detail later in this article.
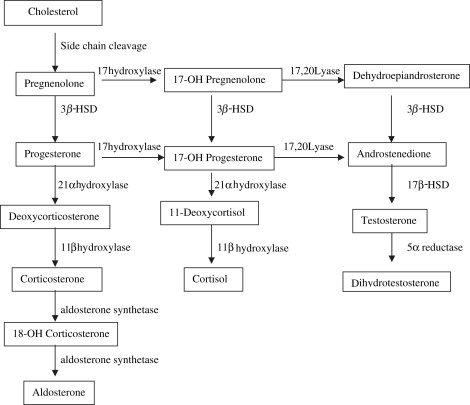
The most common cause of CAH is inactivation of CYP21 , which catalyzes the conversion 17-OH progesterone to 11-deoxycortisol, a precursor of cortisol, and the conversion of progesterone to deoxycorticosterone, a precursor of aldosterone. A spectrum of phenotypes from mild to severe clitoromegaly is possible. Classic 21α-hydroxylase deficiency is comprised of two forms of CAH: a severe, salt-wasting type with a defect in aldosterone biosynthesis and a simple, virilizing type with normal aldosterone synthesis. A mild, non-classic form also exists that can be asymptomatic or associated with signs of postnatal androgen excess. There are two CYP11 genes: CYP11B1 and CYP11B2 . CYP11B1 converts 11-deoxycortisol to cortisol. Alternatively, CYP11B2 converts deoxycorticosterone (DOC) to corticosterone, corticosterone to 18-hydroxycorticosterone, and 18-hydroxycorticosterone to aldosterone. Hypertension, which occurs in about two thirds of patients, is presumptively a consequence of excess DOC, with resultant salt and water retention. Excess androgen secretion in utero masculinizes the external genitalia of the female fetus. After birth, untreated male and female neonates progressively virilize and experience rapid somatic growth and skeletal maturation.
3β2HSD catalyzes three reactions: pregnenolone to progesterone; 17-OH pregnenolone to 17-OH progesterone; and dehydroepiandrosterone (DHEA) into androstenedione. Complete deficiency of 3β2HSD impairs synthesis of adrenal aldosterone and cortisol, and gonadal testosterone and estradiol. These newborns have severe CAH and exhibit signs of mineralocorticoid and glucocorticoid deficiency in the first week of life. Masculinization occurs as a result of DHEA conversion to testosterone in fetal placenta and peripheral tissues manifesting as mild to moderate clitoromegaly.
CYP17 also catalyzes three reactions: pregnenolone to 17-OH pregnenolone, 17-OH pregnenolone to dehydroepiandrosterone, and progesterone to 17-OH progesterone. Phenotypically, affected girls have normal internal and external genitalia, but demonstrate immature sexual development because of an inability of the ovaries to secrete estrogens at puberty. In mild defects, aldosterone secretion may be normal and hypertension absent.
StAR deficiency, also called lipoid adrenal hyperplasia, is a rare form of CAH and represents the most severe genetic defect in steroidogenesis. StAR deficiency is associated with severe glucocorticoid and mineralocorticoid deficiencies because of a failure to transport cholesterol from the outer to the inner mitochondrial membrane, which blocks conversion of cholesterol to pregnenolone. These children are at risk for neonatal demise because of missed adrenal crisis. Neonates suspected to have adrenal insufficiency should be closely monitored for hypoglycemia, hyponatremia, and adrenal insufficiency. Affected girls demonstrate normal internal and external genitalia. Most of these women undergo spontaneous puberty but are at risk for irregular menses, ovarian cysts, and premature menopause.
Ovotesticular disorder of sex development
Ovotesticular DSD requires expression of ovarian and testicular tissue. The most common karyotype in the United States is 46,XX, although 46,XY, mosaicism, or chimerism (46,XX/46,XY) can occur. Although mosaicism may occur from chromosomal nondisjunction, chimerism may result from a double fertilization (an X and a Y sperm) or from fusion of two fertilized eggs. This fairly uncommon condition can be further classified into three groups: lateral ovotesticular DSD has a testis on one side and an ovary on the contralateral (usually left) side; bilateral ovotesticular DSD has an ovotestis on each side; and, most commonly, unilateral ovotesticular DSD has an ovotestis on one side and either a testis or an ovary on the contralateral side. Furthermore, the external genitalia are ambiguous with hypospadias, cryptorchidism, and incomplete fusion of the labioscrotal folds. The genital duct differentiation in these patients generally follows that of the ipsilateral gonad on that side, such as a fallopian tube with an ovary and a vas deferens with a testis.
46, XY disorder of sex development
The 46,XY DSD is a heterogeneous disorder in which testes are present but the internal ducts system or the external genitalia are incompletely masculinized. The phenotype is variable and ranges from completely female external genitalia to the mild male phenotype of isolated hypospadias or cryptorchidism. The 46,XY DSD can be classified into eight basic etiologic categories: (1) Leydig cell failure, (2) testosterone biosynthesis defects, (3) androgen insensitivity syndrome, (4) 5 a -Reductase deficiency, (5) persistent Müllerian duct syndrome, (6) primary testicular failure or vanishing testes syndrome, (7) exogenous insults, and (8) gonadal dysgenesis.
Leydig cell failure
The presence of testosterone, produced by testicular Leydig cells, induces male differentiation of the wolffian ducts and external genitalia. The 46,XY DSD can result from Leydig cell unresponsiveness to human chorionic gonadotrophin hormone (hCG) and luteinizing hormone (LH). The phenotypes of these patients vary from normal female to hypoplastic external male genitalia.
Testosterone biosynthesis enzyme defects
Described earlier in this article for 46,XX DSD, defects in four of the steps of the steroid biosynthetic pathway from cholesterol to testosterone may also produce genital ambiguity in the male. These defects include the less common forms of congenital adrenal hyperplasia: 3β2HSD deficiency, CYP17 deficiency, StAR protein deficiency, and 17bHSD deficiency. Although DHEA conversion into testosterone results in virilization in females, this same process insufficiently masculinizes affected boys. Thus, male infants exhibit ambiguous genitalia with variable degrees of hypospadias, cryptorchidism, penoscrotal transposition, and a blind vaginal pouch. Boys with CYP17 deficiency display a developmental spectrum from the normal female phenotype to the ambiguous hypospadiac male phenotype. The magnitude of the decreased masculinization in the male infant correlates with the severity of the block in 17α-hydroxylation. Affected boys with StAR deficiency have severe testosterone deficiencies and exhibit female external genitalia with a blind vaginal pouch. No surviving patients with 46,XY have demonstrated testis function at puberty. The affected 46,XY boys with 17βHSD deficiency have external female genitalia, inguinal testes, internal male ducts, and a blind vaginal pouch. At puberty, these patients demonstrate an increase in their levels of gonadotropins, androstenedione, estrone and testosterone. Delayed virilization may ensue if some testosterone levels approach the normal range.
Androgen insensitivity syndrome
The broad spectrum of androgen insensitivity syndrome (AIS) ranges from patients with 46,XY with complete AIS, or testicular feminization, to partial AIS. This syndrome is the result of mutations of the steroid-binding domain of the androgen receptor resulting in receptors unable to bind androgens or receptors that bind to androgens but exhibit qualitative abnormalities and do not function properly. This disorder affects 1 in 20,000 live male births with a maternal inheritance pattern, because the androgen receptor gene is located on the long arm of the X chromosome.
The external genitalia of a child with complete androgen insensitivity resembles normal female genitalia although the karyotype is XY and testes are located internally. These children are raised as girls. Most children are not diagnosed until puberty during an evaluation for primary amenorrhea. Occasionally, it is also discovered that at the time of inguinal hernia repair, or more recently, when the prenatal karyotype does not match the external phenotype of the newborn child.
5α-reductase deficiency
5α-reductase deficiency was first described as pseudovaginal perineal scrotal hypospadias. In this autosomal recessive condition, patients have a defect in the conversion of testosterone to dihydrotestosterone (DHT). These patients have a 46,XY karyotype and ambiguous external genitalia but normally differentiated testes with male internal ducts. However, at puberty, significant virilization occurs as testosterone levels increase into the adult male range while dihydrotestosterone remains disproportionately low. There are three genetic isolates of this disorder that have been described: the Dominican Republic, the New Guinea Samba Tribe, and in Turkey. Many of these patients undergo a change of their gender identity from female to male after puberty. Virilization can be secondary to slightly increased plasma DHT levels and to the chronic effect of adult T levels on the androgen receptor.
Persistent Müllerian duct syndrome
Antimüllerian hormone (AMH), or Müllerian inhibitory substance (MIS), is secreted by the Sertoli cells from the time of fetal seminiferous tubule differentiation until puberty. MIS binds to a receptor in the mesenchyme surrounding the Müllerian ducts before 8-weeks gestation causing apoptosis and regression of the Müllerian duct. Because the diagnosis of persistent Müllerian duct syndrome (PMDS) is often made at the time of inguinal hernia repair or orchiopexy, this syndrome is commonly referred to as hernia uteri inguinalis. PMDS can occur from a failure of the testes to synthesize or secrete MIS because of an AMH gene mutation or from a defect in the response of the duct to MIS because of an AMH2 receptor gene mutation. PMDS is inherited in a sex-linked autosomal recessive manner and AMH mutations are most common in Mediterranean or Arab countries with high rates of consanguinity. Most of these familial mutations are homozygous and the patients have low or undetectable levels of serum MIS. In contrast, AMH2 receptor mutations are often heterozygous and are more common in France and Northern Europe. These patients usually have high-normal or elevated MIS concentrations.
Congenital anorchia
Congenital anorchia or vanishing testes syndrome encompasses a spectrum of anomalies resulting from cessation of testicular function. A loss of testes before 8-weeks gestation results in patients with 46,XY with female external and internal genitalia and either no gonads or streak gonads. A loss of testes at 8 to 10 weeks in development leads to ambiguous genitalia and variable ductal development. A loss of testis function after the critical male differentiation period, which is at 12- to 14-weeks gestation, results in a normal male phenotype externally along with anorchia internally. Sporadic and familial forms of anorchia exist. The familial cases, including some reports of monozygotic twins, support the presence of an as yet unidentified mutant gene in some patients with the syndrome.
Exogenous source
Exogenous insults to normal male development include maternal ingestion of progesterone or estrogen or various environmental hazards. As early as 1942, Courrier and Jost demonstrated an antiandrogen effect on the male fetus induced by a synthetic progestagen, and more recently, Silver and colleagues showed an increased incidence of hypospadias in male offspring conceived by in vitro fertilization. They hypothesized that the increased risk may be secondary to maternal progesterone ingestion. Sharpe and Skakkebaek have further postulated that the increase in reproductive abnormalities in men is related to an increase in the in utero exposure to environmental estrogens.
Gonadal dysgenesis
Dysgenetic 46,XY DSD exhibits ambiguous development of the internal genital ducts, the urogenital sinus and the external genitalia. Dysgenetic testes can result from mutations or deletions of any of the genes involved in the testis determination cascade, namely SRY, DAX, WT1 , and SOX9 . The SRY gene is a single exon gene located on the short arm of the Y chromosome. SRY gene mutations usually result in complete gonadal dysgenesis and XY sex reversal or Swyer syndrome. The DSS locus (dosage-sensitive sex reversal) has been mapped to the Xp21 region, which contains the DAX1 gene. Duplication of the DSS locus has been associated with dysgenetic 46,XY DSD. The DSS locus has been theorized to contain a wolffian inhibitory factor, which acts as an inhibitory gene of the testis determination pathway. Swain and colleagues have shown that DAX1 antagonizes SRY action in mammalian sex determination. Male patients with Denys-Drash syndrome have ambiguous genitalia with streak or dysgenetic gonads, progressive nephropathy, and Wilms tumor. Analysis of these patients revealed heterozygous mutations of the Wilms tumor suppressor gene ( WT1 ) at 11p13. The WAGR syndrome (Wilms tumor, aniridia, genitourinary abnormalities, mental retardation) is also associated with WT1 alterations. The urogenital anomalies seen in the WAGR syndrome are usually less severe than in Denys-Drash syndrome. The SOX9 gene has been associated with campomelic dysplasia, an often lethal skeletal malformation with dysgenetic 46,XY DSD. Affected 46,XY children have phenotypic variability from normal boys to normal girls, depending on the function of the gonads.
Swyer syndrome represents an uncommon form of pure gonadal dysgenesis. These children have female external genitalia and have a uterus and fallopian tubes, however, the karyotype is 46,XY with a Y chromosome that usually does not work and two dysgenetic gonads in the abdomen.
Partial gonadal dysgenesis refers to disorders with partial testicular development, including mixed gonadal dysgenesis, dysgenetic male pseudohermaphroditism, and some forms of testicular or ovarian regression. Mixed or partial gonadal dysgenesis (45,XX/46,XY or 46,XY) involves a streak gonad on one side and a testis, often dysgenetic, on the other side. Patients with a Y chromosome in the karyotype are at a higher risk than the general population to develop a tumor in the streak or dysgenetic gonad. Gonadoblastoma, a benign growth, is the most common tumor. Because of the 20% to 25% age-related risk for malignant transformation into a dysgerminoma, surgical removal of the gonad is recommended. Patients with a 45,XX/46,XY karyotype and normal testis biopsy could retain the testis if it is descended or can be placed in the scrotum. These children would then need a close follow-up of the testis by monthly self examinations for tumor formation.
Sex chromosome disorder of sex development
Sex chromosome anomalies comprise another category of DSD. Klinefelter syndrome (47,XXY) usually becomes evident during adolescence as patients develop gynecomastia, variable androgen deficiency, and small atrophic testes with hyalinization of the seminiferous tubules. These patients demonstrate azoospermia and increasing gonadotropin levels. Boys with 47,XXY may develop through nondisjunction of the sex chromosomes during the first or second meiotic division in either parent, or less commonly, through mitotic nondisjunction in the zygote at or after fertilization. These abnormalities almost always occur in parents with normal sex chromosomes. The 46,XY/47,XXY mosaicism is the most common form of the Klinefelter variants. The mosaics, in general, manifest a much milder phenotype than patients with classic Klinefelter. Testes differentiation and a lack of ovarian development in these patients indicates that a single Y chromosome with SRY expression is enough for testis organogenesis and male sex differentiation in the presence of as many as four X chromosomes in some patients with Klinefelter. These testes are not truly normal, however, since they are usually small and azoospermic. Although there are sporadic reports of paternity, most fertile Klinefelter individuals have sex chromosome mosaicism. Pure gonadal dysgenesis (PGD) describes a 46,XX or 46,XY child with streak gonads or more commonly, a child with Turner syndrome (45,XO or 45,XO/46,XX).
46 XX testicular disorder of sex development
Categories of 46,XX testicular DSD include boys with classic XX with apparently normal phenotypes, boys with non-classic XX with some degree of sexual ambiguity, and XX ovotesticular DSD. Eighty percent to ninety percent of boys with 46,XX result from an anomalous Y to X translocation involving the SRY gene during meiosis. In general, the greater the amount of Y-DNA present, the more virilized the phenotype. Although 8% to 20% of boys with XX have no detectable Y sequences, including SRY , about 1 in 20,000 phenotypic boys have a 46,XX karyotype. Most of these patients have ambiguous genitalia, but reports of boys with classic XX without the SRY gene do exist. This phenomenon again raises the possibility of mutation of a downstream wolffian inhibitory factor when cases of normal virilization are seen without the presence of the SRY gene. Some patients with 46,XX testicular DSD have the SRY gene translocated from the Y to the X chromosome. However, for most patients, the genes responsible are not yet identified.
History and Physical Examination
Patients with bilaterally impalpable testes or a unilaterally impalpable testis and hypospadias should be regarded as having DSD until proven otherwise, whether or not the genitalia appear ambiguous. Patient history should include the degree of prematurity, ingestion of exogenous maternal hormones used in assisted reproductive techniques, and maternal use of oral contraceptives during pregnancy. A family history should be obtained to document any urologic abnormalities, neonatal deaths, precocious puberty, amenorrhea, infertility, or consanguinity. Any abnormal virilization or cushingoid appearance of the child’s mother should also be noted. Abnormalities of the prenatal maternal ultrasound are also helpful, such as discordance of the fetal karyotype with the genitalia by sonogram.
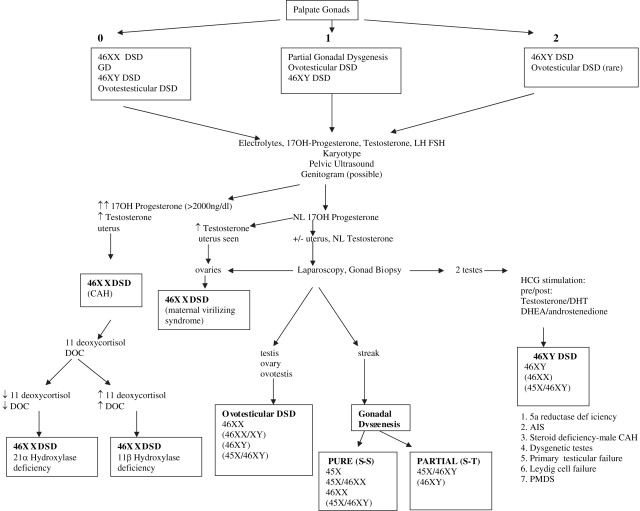
For differential diagnosis and treatment purposes, the most important physical finding is the presence of one or two gonads. If no gonads are palpable, all DSD categories are possible. Of these, 46,XX DSD is most commonly seen followed by 45,X/46,XY. A palpable gonad is highly suggestive of a testis, or rarely, an ovotestis, because ovaries and streak gonads do not descend. If one gonad is palpable, 46,XX DSD is less likely, whereas 45,X/46,XY, ovotesticular DSD, and 46,XY remain possibilities. If two gonads are palpable, 46,XY, and rarely ovotesticular DSD, are the most likely diagnoses ( Fig. 2 ).
Related posts:
 Hypospadias: Etiology and Current Research
Hypospadias: Etiology and Current Research
 Cryptorchidism: Pathogenesis, Diagnosis, Treatment and Prognosis
Urologic Care of the Neurogenic Bladder in Children
Contemporary Surgical Management of Pediatric Urolithiasis
Treatment Strategy for the Adolescent Varicocele
Cryptorchidism: Pathogenesis, Diagnosis, Treatment and Prognosis
Urologic Care of the Neurogenic Bladder in Children
Contemporary Surgical Management of Pediatric Urolithiasis
Treatment Strategy for the Adolescent Varicocele
 Laparoscopic and Robotic Approach to Genitourinary Anomalies in Children
Laparoscopic and Robotic Approach to Genitourinary Anomalies in Children
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


