Overall Bottom Line
- The term LFTs is often misused to refer to serum chemistry tests.
- Liver disease is evaluated by tests that detect liver injury, impaired bile flow or cholestasis, synthetic capacity, excretory function and metabolic function.
- Interpretation of abnormalities of the specific tests in the context of pediatric liver disease is described.
- Screening tests that need to be performed when abnormal aminotransferases are noted are described.
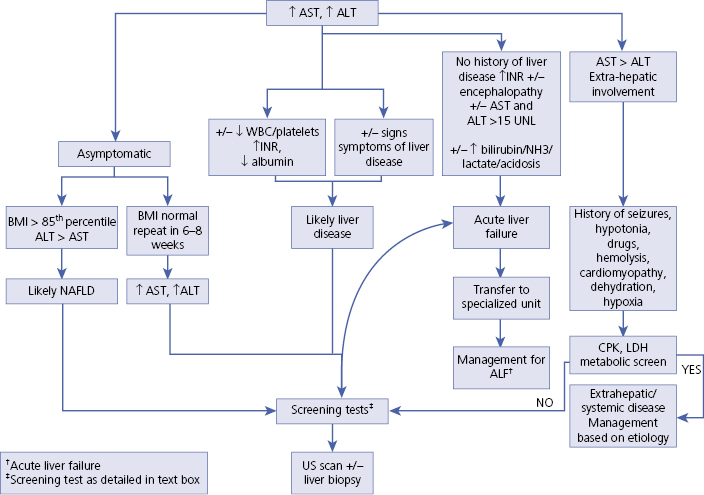
- An algorithm suggesting an approach when a child presents with abnormal aminotransferases is presented.
Section 1: Tests that Evaluate Liver Injury (Algorithm 35.1)
- Serum aminotransferases include ALT and AST.
- LDH is a cytoplasmic enzyme present in many tissues including liver and therefore has limited specificity.
- ALT (formerly SGPT) is primarily localized to liver and in the cytosol.
- AST (formerly SGOT) is present in liver (mitochondria and cytosol), heart, skeletal muscle, kidney and brain.
- The aminotransferases lack some sensitivity as values may be normal despite presence of inflammation on liver biopsy and also lack specificity as they may be elevated in non-hepatic conditions like myopathy and hypothyroidism.
- The ULN for ALT varies widely and many use a value around 50 IU/L. More recently, a study using National Health and Nutrition Examination Survey data showed that the 95th percentile levels for ALT in healthy weight, metabolically normal, liver disease-free, pediatric participants were 25.8 IU/L (boys) and 22.1 IU/L (girls), much lower than is used in clinical practice.
- The most common cause of elevated aminotransferases in pediatric practice is NAFLD; BMI >85th percentile overweight, BMI >95th percentile obese.
– – – – – – – – – –
Algorithm 35.1 Diagnosis of abnormal transaminases in children
– – – – – – – – – –
Elevated ALT and AST commonly seen in children
- NAFLD.
- Chronic liver disease:
- Viral hepatitis.
- Autoimmune hepatitis.
- Wilson disease.
- Cholestatic liver disease.
- Cryptogenic cirrhosis.
- Viral hepatitis.
AST and ALT >15 times normal
- Acetaminophen/toxin induced.
- Hypoxia/hypoperfusion – “shock liver”.
- Acute viral hepatitis.
AST > ALT
- Hemolysis.
- Acute rhabdomyolysis – viral illness/vigorous physical activity.
- Myopathy.
- Myocardial disease.
- Macro AST.
ALT > AST
- NAFLD.
- Celiac disease.
Screening laboratory tests for abnormal aminotransferases
- CBC, INR.
- BUN, creatinine, electrolytes, albumin, GGT, total and direct bilirubin.
- Toxicology screen, glucose, ammonia (when ALT, AST >10 times normal).
- Viral hepatitis: hepatitis A IgG and IgM, hepatitis B surface antigen and antibody, hepatitis C antibody.
- Autoimmune hepatitis screen: immunoglobulin G, antinuclear antibody, smooth muscle antibody, liver kidney microsomal (LKM) antibody.
- Wilson disease screen: ceruloplasmin, 24 hour urinary copper.
- Alpha-1 antitrypsin deficiency: alpha-1 phenotype.
- Creatinine phosphokinase, aldolase, LDH (AST > ALT, concern for extra-hepatic cause of elevated aminotransferases).
Section 2: Tests that Evaluate Impaired Bile Flow or Cholestasis
- ALP.
- GGT.
- 5’nucleotidase.
Serum GGT
- Serum GGT varies by age: normal value up to 400 IU/L in neonates and reducing to 50 IU/L by a year of age.
- GGT is elevated typically in biliary obstruction.
- Cholestasis with low/normal GGT is characteristically seen in progressive familial intrahepatic cholestasis (PFIC 1 and 2) and inborn errors of bile acid synthesis.
Increased GGT
- Biliary atresia.
- Choledochal cyst.
- Biliary stones/stricture.
- Inspissated bile syndrome.
- Sclerosing cholangitis.
- Alagille syndrome.
- Alpha-1 antitrypsin deficiency.
- Anticonvulsants like phenobarbitone, phenytoin.
ALP
- The most likely source of ALP are liver and bone.
- Normal growing children and adolescents have elevation of ALP of bone origin.
- Increased ALP associated with elevated GGT or 5’nucleotidase is considered of hepatic origin.
- Increased ALP in the absence of liver disease may be:
- Secondary to chronic renal failure.
- Familial inheritance, blood type B or O.
- Pregnancy.
- Transient hyperphosphatemia of infancy.
- Secondary to chronic renal failure.
- Decreased ALP is seen in:
- Zinc deficiency (zinc is a co-factor for ALP).
- Wilson disease.
- Zinc deficiency (zinc is a co-factor for ALP).
PFIC
- Is a rare but important familial cause of cholestasis in the newborn.
- Is divided into three types based on mutations in genes encoding transport proteins responsible for the production of bile.
- Mothers of children with PFIC, who are heterozygous for the mutation especially ABCB4 have intrahepatic cholestasis of pregnancy, pruritus during pregnancy, post-natal resolution.
PFIC1 (Byler disease)
- Mutation in ATP8B1, a P-type ATPase that is an aminophospholipid flippase that is responsible for maintaining canalicular lipid asymmetry.
- Presents by 1 year of age with low GGT cholestasis, elevated transaminases, pruritus, hepatosplenomegaly, malabsorption, failure to thrive and rickets.
- Given the protein is present in other epithelia – there is also extrahepatic involvement with diarrhea, deafness, pancreatitis, renal tubular acidosis and/or lung disease.
- Histology reveals a bland hepatocellular, canalicular cholestasis with coarse granular bile on electron microscopy.
- Biliary diversion may help the pruritus. Liver transplantation is not the preferred option in view of extra-hepatic involvement.
PFIC2
- Mutation in ABCB11 which codes for bile salt export pump that is responsible for bile acid transport across the canalicular membrane.
- Similar to PFIC1 there is low GGT cholestasis in infancy, with transaminases elevated >5 ULN, but there is no extra-hepatic involvement.
- Rapid progress to cirrhosis and possibility of development of HCC and cholangiocarcinoma in the first year of life.
- Histology shows giant-cell hepatitis and lobular cholestasis with finely granular/filamentous bile in dilated bile canaliculi on electron microscopy.
- Monitoring of AFP and regular US scans important.
- Biliary diversion and liver transplantation are required in many.
PFIC3
- Mutation in ABCB4, which codes for MDR3 (multidrug resistance protein 3), causing malfunction of a transporter required for biliary phosphatidylcholine secretion → reduced biliary phospholipid → renders the biliary epithelium susceptible to damage by the biliary bile acids.
- Age of onset is variable but usually after infancy and GGT is high.
- Histology shows bile ductular proliferation and mixed inflammatory infiltrates. Periductal sclerosis affecting the interlobular bile ducts and biliary cirrhosis eventually occurs.
- Oral ursodeoxycholic acid is useful.
- Liver transplantation is required once cirrhosis develops.
Benign recurrent intrahepatic cholestasis (BRIC)
- Milder form of low GGT cholestasis.
- BRIC1 and BRIC2 are thought to be secondary to missense mutations of ATP8B1 and ABCB11 respectively.
- Intermittent episodes of cholestasis and severe pruritus.
- Chronic liver damage does not occur.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


