Overall Bottom Line
- WD, also known as hepatolenticular degeneration, is a rare autosomal recessive systemic disorder caused by defective copper metabolism.
- It most commonly presents with isolated hepatic, neurological or psychiatric manifestations but multisystem involvement is not uncommon.
- Diagnosis of WD involves characteristic clinical, biochemical and molecular findings.
- Combined treatment with chelators, zinc and liver transplantation has proven lifesaving and even curative.
- Potential future therapies include isolated hepatocyte transplantation and gene therapy.
Section 1: Background
Definition of disease
- WD is an inherited rare autosomal recessive disease of copper metabolism characterized by copper accumulation in hepatocytes and in other extra hepatic organs.
Prevalence
- The prevalence of WD is estimated at three per 100 000.
- Siblings of a WD patient have a 1 in 4 risk of disease; children of a WD patient have one in 200 risk.
- The frequency of carriers of the ATP7B mutation is about 0.6–1%.
- Consanguinity increases prevalence in some regions (e.g. Crete, Sardinia, Japanese islands).
Economic impact
- Given the low prevalence of WD, its economic impact is not great.
- WD and alpha-1 antitrypsin deficiency disease combined account for less than 5% of liver transplant procedures in the USA according to Organ Procurement and Transplantation Network data.
Etiology
- Homozygous or compound heterozygous mutations in the ATP7B gene (locus: 13q14.3-q21.1), which codes for an ATP-dependent copper export pump, are the cause of WD.
- Up to 300 mutations have been associated with WD.
Pathophysiology
- Copper is absorbed via the small intestine into the portal circulation. In the liver, hepatocytes take up copper and incorporate the copper into ceruloplasmin. Excess copper is bound to Apo metallothionein or secreted into bile.
- In WD due to mutations in the ATP7B gene, the incorporation of copper into ceruloplasmin and excretion of excess copper into bile is impaired.
- The excess copper is retained in the hepatocytes where it promotes free radical formation and oxidative damage to cellular lipids and proteins.
- As the hepatic copper burden increases further, copper is released into the circulation and deposited in other organs such as the nervous system.
- Secretion of ceruloplasmin into the circulation is dependent on copper incorporation. Hence serum ceruloplasmin levels are low in WD.
- Copper deposition in the cornea may manifest as Kayser–Fleischer (KF) rings.
Predictive/risk factors
- Inheritance of dual mutations in the ATP7B gene – nearly 100% penetrance.
Section 2: Prevention
- Low copper diet and zinc or chelation therapy may prevent disease onset.
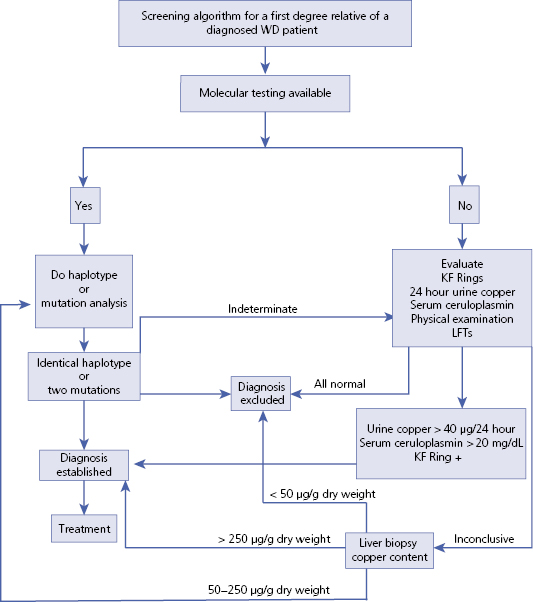
Screening (Algorithm 16.1)
- Genetic, ophthalmologic and biochemical tests are used to screen for WD. It is not cost effective to screen the general population due to the large number of known mutations and the rarity of the disease. However, first degree relatives of any patient with WD should be screened since early treatment can prevent disease onset.
- Specific testing can be postponed until an abnormality develops on a regular check up or blood tests.
- For infants of affected families, a delay in achieving a developmental milestone should lead to testing.
– – – – – – – – – –
Algorithm 16.1 Screening algorithm for Wilson disease
– – – – – – – – – –
Primary prevention
- Low copper diet and zinc or chelation therapy may prevent disease onset.
Secondary prevention
- Liver transplantation will prevent disease recurrence.
Section 3: Diagnosis (Algorithm 16.2)
Clinical Pearls
- WD should always be considered in a patient <40 years of age who presents with unexplained liver, neurological or neuropsychiatric disease.
- Its clinical spectrum ranges from asymptomatic hepatomegaly or minor liver enzyme derangement to fulminant liver failure with neuropsychiatric symptoms.
- AP levels may be relatively low in WD.
- Presence of low serum ceruloplasmin levels, increased urine copper excretion and KF rings help in diagnosis of WD.
- If clinical and biochemical tests are inconclusive then confirmation can be performed by quantification of copper in the liver via liver biopsy or by molecular techniques such as haplotype or mutation analysis.
Differential diagnosis
| Differential diagnosis | Features |
|---|---|
| Drug or toxin-induced hepatitis/liver failure | A thorough history is needed to rule out hepatotoxicity due to medications and toxin exposure. Be aware that multiple products contain acetaminophen |
| Viral hepatitis | History of travel, transfusion, needle sharing, or tattoos. Positive viral serological markers |
| AIH | Characterized by serum autoantibodies and interface hepatitis on the biopsy |
| NAFLD | Normal serum ceruloplasmin and urinary copper levels. No KF rings |
| Other psychiatric disorder | Normal serum ceruloplasmin and urinary copper levels. No KF rings. Diagnose by DSM IV |
Typical presentation
- The most common age for presentation ranges from 5 to 40 years with hepatic features mostly preceding the neurological features except for those diagnosed in late second or third decade.
- The clinical spectrum of liver disease varies from asymptomatic liver enzyme elevation to chronic active hepatitis and cirrhosis or may present with acute liver failure with the most common initial presentation being cirrhosis. Those presenting with acute liver failure may also have hemolytic anemia and acute renal failure.
- Initial neurological manifestation is generally asymmetric tremors but other manifestations such as dysphagia, ataxia and mask-like facies are fairly common. About a third of patients manifest a neuropsychiatric finding: emotional liability, anxiety or even psychosis.
Clinical diagnosis
History
- It is important to enquire about symptoms of chronic liver disease and cirrhosis such as fatigue, confusion, GI bleeding, easy bruising and swelling.
- In fulminant liver failure cases, always exclude a history of drug intake (e.g. acetaminophen) and autoimmune hepatitis before diagnosing WD.
- Neuropsychiatric manifestations like tremors, mask-like facies, depression, mood swings should also be reviewed.
- New difficulties in school may presage WD onset in children.
- Always ask about risk factors (e.g. injection drug use or alcohol abuse) for other liver diseases and a family history of liver and neuropsychiatric disease.
Physical examination
- Evaluate for signs of chronic liver disease and cirrhosis.
- Refer to neurologist and ophthalmologist for detection of subtle neurological and ophthalmological (i.e. KF rings) findings. KF rings are present in 44–62% of patients.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


