Fig. 13.1
Investigational New Drug (IND) vial-ampoule manufacturing machine Clinical Scale vialampoule aseptic processing isolator system
13.2.1 Quality Control of Sterile Drugs
When validation was first introduced in the 1970s, the general concept driving the effort was that “quality control testing alone can’t secure quality.” For example, in sterility test, detection rate is very low because only a limited number of product samples are tested. The number tested, most commonly 20 articles, is not enough to guarantee the sterility of product lots that may consist of 10,000–100,000 or more units. If sterility test uses the samples of 10 ml at 10 locations from each lot, the microbial detection rate of 1 ml is less than 0.0105 unit, which means microorganisms cannot be detected unless there is more than one microorganism in 100 ml [2]. This likelihood of contamination shown in this example would only be true if the limit of detection of the method is one cell, which is very unlikely to be the case. Thus, the sterility test falls far short of being able to guarantee the sterility of products. From this viewpoint, a validation approach focusing on the manufacturing facility and aseptic process control strategies is required to ensure lot-to-lot product safety. Furthermore, the current philosophy of the International Congress for Harmonization is one based upon Quality by Design (QbD) This approach to process design means that consideration of microbial contamination control must begin very early in the process by which a new facility is designed or by which a new product is added to an existing manufacturing system. In other words, a facility and process must be carefully designed within the defined design space to ensure product safety under all possible operating conditions. In terms of microbiological safety, this means designing so that the probability of microbial contamination is minimized to the greatest extent possible.
13.2.2 Risk and Sterility
Risk management in aseptic processing consists of two different critical factors. The first of these is “risk evaluation/analysis,” the goal of which is risk quantification. The other is “risk mitigation” that considers possibility of contamination during facility design, equipment selection, definition of process, and the operation of process itself. Certainly at the present time, risk evaluation is a significant focus within industry, but risk mitigation is by far the most important of the two factors listed above. Risk mitigation can both indicate the level of need for process improvement and determine the priority that should be assigned to mitigation activities. Risk mitigation as it relates to aseptic processing generally means to properly control human intervention, but ultimately if risk mitigation is to be maximized, personnel must be completely removed from critical area of aseptic processing.
13.2.3 Effectiveness of Gowns
It continues to be widely assumed by some scientists and engineers that aseptic conditions can be adequately maintained if the personnel conducting aseptic processing wear aseptic gowns properly and exhibit sound aseptic technique in conducting their work. However, it is clear from published research data that personnel emit very high levels of microbiological contamination into the environment. This is true even when state-of-the-art gowns are chosen and worn properly. According to Ljungqvist et al., personnel wearing previously unused sterilized gowns emit 1500–3000 cfu/h of microbiological contamination even when conducting limited operations [3]. The microbiological contamination released by personnel can increase to ≥10,000 cfu/h during complex operations that require the execution of many physical tasks.
The total number of aerobic bacteria existing on human skin is normally >1.2 million per square meter. The number of microorganisms existing on a healthy human’s hands and arms are in the range 0.9–3 million per square meter [4]. In case of anaerobic bacteria, such as P. acnes, the number of organisms present can be several times more than the quantities shown. Microorganisms are emitted by gowned personnel by a pumping effect of the technician’s gown (air coming in and out from, for instance, operator’s chest depending on the task) during operation. In recent years, advanced technology has been introduced to reduce the possibility of product contamination caused by personnel in manufacturing site of sterile drugs. These technologies often known collectively as advanced aseptic processing technologies include Blow-Fill-Seal and Restricted Access Barrier System, in addition to isolators.
13.3 Isolator Technology
13.3.1 What Is an Isolator?
Isolator is defined as a system having an aseptic processing area that is both physically separated from the environment and does not permit direct interventions by personnel. The system generally has continuous supply of fresh or recirculated air through HEPA or ULPA (ultra-low particulate air) filters. This filtered air supply is maintained at a flow rate that results in a positive air pressure in the isolator being maintained relative to the surrounding environment. Isolators that have a direct opening to the surrounding environment are said to be aerodynamically sealed or sealed by air overspill. This simple concept of aerodynamic separation is a well-proven principle, which has been demonstrated to work well. However, many isolators operate without a direct opening to the external environment [5].
Isolator systems do not allow direct interventions by personnel into the enclosure’s aseptic processing area. To ensure that contamination is not introduced into the isolator, it is critical to have means to bring materials into and out of the isolator in a way that presents the transfer of contamination into the isolator. The sterility assurance provided by an isolator is a product of physical separation of personnel, elimination of risk of introduction of airborne contamination, and systems that prevent the introduction of non-sterile materials into the isolator. Safe, contamination-free transfer of materials into and out of the isolator can be assured by the use of devices known as decontamination pass boxes and circular-shaped rapid transfer ports (RTP); also decontamination interfaces for attaching devices to isolators have been developed (Fig. 13.2).
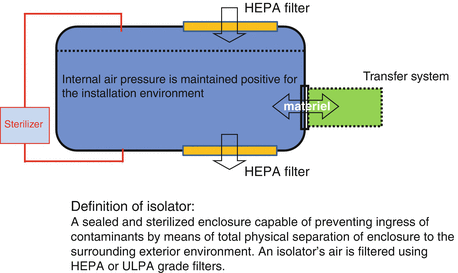
Fig. 13.2
What is an isolator?
13.3.2 General Features of an Isolator
Aseptic processing that relies on the use of a conventional clean booth or a clean room approach is always at risk from contamination contributed by human operators and technicians. This unavoidable fact makes contamination control difficult. In reality, effective countermeasures against contamination in conventional clean booth bring with them massive increases in facility and operational costs. In contrast, the most significant advantage of an isolator system is the comparative ease with which an aseptic environment can be maintained. Isolators have the added advantage of minimizing the actual volume of clean space that must be maintained in an essentially sterile manner. This allows a dramatic increase in operating costs through reduced energy consumption and the elimination of a need for consumables such as aseptic gowns.
ISO 5/Grade A environments are required for the critical area of aseptic processing. The critical area in aseptic processing is defined as the part of a facility where product and sterilized components are brought together and filled and/or assembled. It is necessary to install and operate this ISO 5/Grade A critical zone environment within an ISO 7/Grade B environment and, of course, to provide proper gowning and support facilities to ensure safe, contamination-free personnel and component entry. Alternatively, in some modern facilities, the entire aseptic processing room can be designed to ISO 5/Grade A requirements; however, such a facility is both expensive to build and also to operate.
In contrast, an isolator for aseptic processing of pharmaceuticals is installed in a much less expensive ISO 8/Grade D environment; therefore, the strict clean room-style aseptic gowning of personnel is not necessary [1].
Additionally, the surface area in clean rooms is typically about twice as large as that of isolators [6]. Because an isolator is a microbiologically closed environment which can be efficiently treated with sporicidal agents, the inherent risk of microbial contamination is extremely low. The essentially sterile condition inside the isolator is readily maintained by design features such as air filtration and overpressure, which prevent the ingress of contaminations from the surrounding environment. According to authors’ experience, the sterile condition inside an isolator can be maintained for one month or more, based on microbial recovery studies done inside an operational isolator using active air samplers and passive air samplers such as settle plates [5]. In contrast, in unidirectional air flow cabinets (clean booths) or manned clean room facilities, product contamination is prevented by maintaining asepsis using unidirectional flow under HEPA filter and avoiding human intervention in critical area as much as possible. Thus, sterility assurance is depending upon personnel training and maintaining strict control of both air flow and work flow of personnel throughout the workspace.
A potential disadvantage of isolators is reduced productivity owing to restricted or inconvenient work access using gloves. Therefore, it is essential to design and fabricate the isolator system with careful attention to ergonomics. It is always best for complex operations to fabricate mock-up isolators in which human interactions with critical equipment through isolator gloves can be evaluated prior to construction. It can be very difficult to correct ergonomic deficiencies in the field after an isolator is built and installed. With this fact in mind, a careful evaluation of ergonomics often using a mock-up isolator fabricated from wood panels or framework is a necessary design step. Our experience has taught us that compromising on operator access and comfort in the design of an isolator is never a good idea. It can result in designs that are unsafe both to the operator and to the product.
13.3.3 The Isolator: A Very Brief History
Around the end of the 1950s, devices that have similar characteristics to a modern-day pharmaceutical isolator had already been used in European nuclear power industry to protect personnel from risk associated with handling radiation and high toxic materials, such as plutonium. The main function of these isolators was protection of personnel. Later, the technology was utilized in laboratories in which studies were conducted on pathogenic microorganisms, and as previously discussed isolators were then used in the healthcare manufacturing industry in analytical microbiology laboratories for sterility testing.
The first aseptic processing isolator was installed at a European pharmaceutical firm around the end of the 1970s for the purpose of sterility test. In 1981, the second isolator was installed, consequently decreasing false-positives due to personnel significantly, boosting attention of its use for sterility test.
The isolator for manufacturing sterile drugs was first installed at Baxter’s Round Lake Factory (United States) and Valencia Factory (Spain), followed by SyntheLabo, Merck, and Novartis. The installation of isolators began to increase more rapidly around 1990, although it can be argued that regulatory compliance uncertainties slowed the pace of implementation until the early years of the twenty-first century.
In Japan, Shibuya Kogyo started to sell isolators in 1992 and installed the first system in Japan in 1994. Then in 1995, the company installed one of the largest and most complex aseptic production isolator systems in the world at a pharmaceutical firm in western Japan. Since that time isolator technology has become widespread in its use in Japan and throughout the developed world.
13.4 Development of Room Decontamination Technology
One of the key operational features of isolators is the ability to create an essentially sterile environment within the enclosure. Both gas and vapor phase sporicidal decontamination agents (sterilants) have been used for such to eliminate bioburden from the isolator enclosure. It is necessary that agents used to decontaminate isolators be both effective sporicides and capable of being used at or very near room temperature. It is important that agents used to decontaminate an isolator desorb readily from the isolator enclosure, equipment, and contents. It is also important that a decontamination agent has a manageable safety profile and that it also is not prone to producing toxic residues. The need for decontamination to be done at generally low temperatures and yet operate quickly and without toxicity has limited the available choices of suitable agents.
13.4.1 Room Decontaminant and Disinfectant
Ethylene oxide was considered a candidate sterilant in the early days of isolator technology but was quickly ruled out because of its toxicity and various safety concerns. Ethylene oxide is both a carcinogenic and mutagenic compound. Formaldehyde was also considered for use with isolators; however, it is also now known to have serious toxicity issues; it is an alkylating agent classified by the IARC as a Group 1 carcinogen. Attempts have been made to use ozone for the purpose of both isolator decontamination and clean room disinfection. Ozone is a powerful oxidant which is a strong sporicide given a high enough concentration and sufficient humidity. Ozone also has the advantage of decomposing rapidly into O2; however, it also causes system design problems because it is highly corrosive and can adversely affect materials used to construct equipment that is often located inside isolators including metals subject to oxidation.
The agent currently available that has the best set of properties overall for isolator decontamination is hydrogen peroxide which can be utilized either as a fine mist or as a vapor. Hydrogen peroxide is a compound that degrades into O2 and H2O, a reaction that can be facilitated by a mixed bed catalyst. Most plastics adsorb H2O2 vapor to only a limited degree and outgas rather quickly during aeration using fresh air. Many metals and glass do not adsorb H2O2, and since these are common materials of isolator construction, this means reasonable decontamination process times of about 3–8 h can be readily attained. H2O2 is not without problems as a decontamination agent; it is corrosive at high concentrations and must be handled and stored properly.
13.4.2 Decontamination by Vapor Phase Hydrogen Peroxide (VPHP)
Sporicidal ability of VPHP was discovered in the late 1970s. The initial research and development was focused on its use as a decontaminating agent of facilities and equipment of hospitals. In the late 1980s, as isolator technology started to emerge in the pharmaceutical and biopharmaceutical industry, experiments were done to determine the suitability of VPHP for this application [7]. Once successes were reported, VPHP began to be used as the primary method for isolator decontamination.
Related posts:
 Critical Path Initiative for Regenerative Medicine in Japan
Regulation for Gene and Cell Therapy Medicinal Products in Europe
Critical Path Initiative for Regenerative Medicine in Japan
Regulation for Gene and Cell Therapy Medicinal Products in Europe
 Liver Regeneration Therapy Using Autologous Bone Marrow-Derived Cells for Cirrhotic Patients
Liver Regeneration Therapy Using Autologous Bone Marrow-Derived Cells for Cirrhotic Patients
 Cell-Based Immunotherapy for HCC: Our Experiences and Future Directions
Cell-Based Immunotherapy for HCC: Our Experiences and Future Directions
 Novel Immunotherapy Using Liver-Derived Natural Killer Cells for Preventing Hepatocellular Carcinoma Recurrence in Liver Transplantation
Novel Immunotherapy Using Liver-Derived Natural Killer Cells for Preventing Hepatocellular Carcinoma Recurrence in Liver Transplantation
 Cell and Vector Production Facility for Gene Therapy and Cell Therapy
Cell and Vector Production Facility for Gene Therapy and Cell Therapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


